I just try it again, not change the method.
@jpfeuffer Now I have a question about that I have the files, but it doesn’t fit the workflow. If I choose the .fasta, The OMSSAAdapter is red, If I choose the .psq, The PeptideIndexer is red. I think because of the files. The right file is the picture.


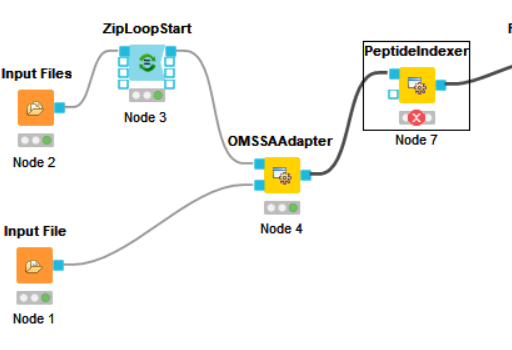
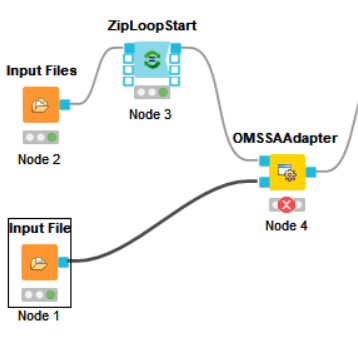
I think all files need to be present and they all need to share the same filename.
Input should always be the .fasta
Yeah, I change the filename and this is wright.
This topic was automatically closed 90 days after the last reply. New replies are no longer allowed.