I would like to calculate RMSD values of 3D coordinates of a small molecule / fragment versus larger compounds containing the fragment as a substructure.
more in detail:
let’s say I have a list of molecules with both SMILES as a column and 3D coordinates in SDF as a column. These are results of a docking workflow. That means the column contains many poses of different molecules, but also several poses from the same molecule. All these molecules are about 300-450 Dalton and contain a common substructure that is the size of a small fragment, 120-200 Dalton. Also for this substructure, the fragment, I have 3D coordinates as SDF, found experimentally via X-ray crystallographic screening.
What I want to know:
how big is the 3D RMSD of the fragment vs the same atoms inside the bigger molecule?
In order to use that for further filtering, i.e. which docking poses moved far away from the input fragment coordinates.
my unsuccessful tries so far:
As far as I have tried the RDkit RMSD filter cannot do it, as
a) I cannot give a reference molecule
b) computes the RMSD between the entire molecule as far as I tested
(thus I would have to cut back the large molecule to the fragment (=MCS), but how to do that in KNIME? )
secondly I tried the KNIME the 3D RMSD node of CDK,
similar problems as with the other, plus
c) does not seem to give pair-wise RMSD but just overall RMSD in the whole column
I have a machine with 64 CPU (128 threads) and it should work on about 1 million poses or so in a reasonable time (e.g. 2h).
So as long as the solution can be parallelized, should be fine.
Has someone here maybe already dealt with a similar problem before and can help me, or point me in some directions?
I know it is a quite detailed problem (at least for us), but any tip is very much appreciated.
I’m not really aware of a way of doing this directly at the moment, however, it ought to be possible to achieve this in one of 3 ways:
Using the RDKit Python API within a python snippet
Using the RDKit Java API within a Java snippet (there is some discussion of how to access the RDKit API in this manner here and here)
Writing a custom node based on the RDKit API
(These are at least vaguely in order of increasing deviation away from most people experience of using RDKit and/or KNIME!)
Most of the bits required are performed one way or another by our Templated Conformer Generation node (see https://kni.me/n/wK3RJiystQYq5M9w), but not in a way that you can access directly. The source code might, however, give some clues as to how to implement using the Java API.
I think the key steps you would need are:
Find the MCS
‘Paste’ the coordinates of the MCS match from each half of the pair onto 2 copies of the MCS structure (or delete all non-matching atoms from each structure)
I was looking a bit more into the MCS node of RDKit, but unfortunately could not find the coordinates info in the output. I have to say that I am not a programmer, so creating my own nodes or writing something myself in Java/Python is simply out of reach for me.
Thought that maybe it could be solved with a complex/creative combination of nodes or so. Hopefully more people would benefit from a solution to my problem, so that it might be integrated in some future plans of the developers.
From the 3 key steps you suggested, maybe one would not even need to find the MCS, as all my molecules definetly contain all the heavy atoms of my reference molecule. So somehow it could be enough to “highlight” the corresponding atoms inside the query molecules and get their coordinates. From that one could create the “sub-molecule” in SDF and then use this for comparing to the reference via the 3D-RSMD node that already exists. … but again, just a therorethical idea. I don’t know how to do it, neither in KNIME nor via other tools.
The MCS is a substructure match, so doesnt contain co-ordinates info in itself. If one of your molecules contains all the atoms of the other every time then the example workflow attached should work. RDKit RMSD.knwf (14.0 KB)
It looks through all the possible matches from a ‘reference’ column to a ‘test’ column and finds the best RMS between them (using some code copy/pasted from the above source code link)
thank you so much for preparing this. That looks really great.
So far, I could get your example to work.
If I load in a large table of mine, it still runs fine (and very quick ), but when I then want to look at the output of the cross-joiner or the Java snippet node, then my GUI crashes … probably something specific to my system, so I opened a ticked with the support. As soon as this is resolved, I will try it out more.
Just to already make sure I use it correctly:
In my case I have docking poses in SDF format, so I will use the “RDKit to Molecule” node to create an RDKit Molecule column that I then feed into the crossjoiner, correct?
Yes - that sounds right. If you have different molecules, then rather than the cross joiner, you will need to use a normal joiner node and join the reference structure for each molecule to the rest of the conformations. This might be enought to sort out the crashes too.
I’ve seen crashes before in the node table views when there are lots of structures. There are a few things you can do to help:
1 - Process the input data in chunks in a chunk loop of say 10,000 rows
2 - Increase the memory available to KNIME by editing the knime.ini file in the installation folder (there are instructions at https://www.knime.com/blog/optimizing-knime-workflows-for-performance for how to do that)
3 - Carry on regardless - If you can filter the structure columns after the java snippet then you might be back ok
even for just 100 rows and 10 columns the table viewing crashes the GUI, also increasing memory extensively did not help. (with 6 rows it’s OK, strangely) Actually, filtering out the RDkit column as you suggested helps, so maybe these are the troublemakers or some of my molecule inputs are bad and then lead to strange RDkit cells which in turn cause the problem. As the command line writes out some Java pointer error, it might also be connected to my specific installation/computer.
Anyway, the great support people of KNIME are on it, until then, filtering out the RDkit columns is a good workaround.
Unfortunately, the RMS that is calculated by the Java Snipped is not what I was looking for.
I created a little set of molecules to test the workflow in the following way:
I used a template molecule and then I created little variations of it my moving/turning/tilting the template differently in Pymol and again exporting as SDF. But unfortunately, for all these molecules (where the atoms are then identical by design, but with moved coordinates), the Java Snipped calculates an RMS = 0.
I also added some molecules that are larger, but contain all the heavy atoms of the template. There I get RMS values between 1.2 and 1.5, but they do not correspond to the actual extend of how much the MCS part moved in reference to the template.
not to bother you further, just in case you or more people are still interested in this problem, I uploaded this little set here:
(just a few molecules, so very slim SDF files)
templatemolecule
testmolecules incl one template molecule as control for “RMS=0”
Ah - so the code as it stands is calculating the best alignment transform to get the smallest RMSD between the two molecules when they are optimally overlaid. Maybe what you want is the distance between the centroids of the atoms they have in common?
I am not sure if this would be an adequate replacement for the RMSD, but maybe could work as well to judge how far the test molecules moved in respect to the template. Would that be easy to derive in your experience?
Could it then be that this would come with a slight downside of not working so well for turns/tilts of the two MCSs in respect to each other, as the MCS centroids could still stay in the same place when the molecules turn/tilt, or not?
But even if this might be a drawback, it would still be great to be able to exclude largely moved molecules.
In the begining I was aiming for an deviation/difference of the individual MCS atoms of both molecules (template vs test) in absolute space. For example, if you open identical but moved molecules from SDF files in Pymol or suchlike, they will appear at different positions in the absolute space.
Meaning the SDF seems, in addition to the info of how the atoms are located relative to each other withing the molecule, to also contain some absolute coordinates. And that, in some way, could be used to calculate the differences. At least that was my theoretical idea from a rookie and non-programmer perspective.
But that seems to involve two separate problems:
mapping the MCS atoms to each other, i.e. which template atom corresponds to which test molecule atom, which the Java code
what are the distances between these atom pairs and maybe add them up as a RMSD
… maybe centroids is an easier option to start with?
best regards,
Jan
PS:
with a bit of google-ing I found that there is an RDkit function called
rdkit.Chem.rdMolAlign.GetAlignmentTransform
that also returns a transformation matrix, if I understood correctly.
But I have no idea how this then could be transformed into a value that describes the distance/difference and that one could use for filtering…
Ah, of course - the rotations mean you do need RMSD. That should be possible to calculate from the two molecules. I’m at the RDKit User Group for the next few days - I’ll have a think how to do it.
with a bit of google-ing I found that there is an RDkit function called
rdkit.Chem.rdMolAlign.GetAlignmentTransform
that also returns a transformation matrix, if I understood correctly.
You do understand correctly - that is how it currently calculates the RMSD (buried in the Java Snippet), but that is for the optimally aligned pair rather than the pair in their current positions.
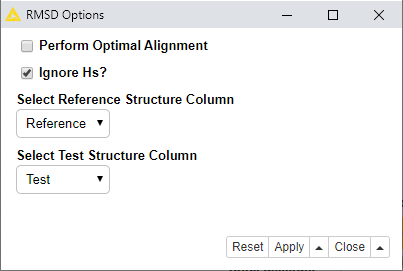
There is a component/wrapped metanode with some options which you might want to tweak. The defaults are as shown:
If you don’t check the ‘Perform Optimal Alignment’ option, then it will work through every combination of substructure match against reference, and calculate the RMSD for each match, and then take the smallest resulting value.
Steve, this is awesome!
works very nicely, thank you so much. That really solves a big problem for me.
Nice of you to also initiate the feature request. I am quite sure this is a very useful solution for many people designing compounds on the basis of crystallographic data.
I hope you don’t mind a little follow-up question:
will this work as perfectly if the Reference and Test molecule in principle have the same atoms as MCS, but in a tautomeric form?
I would guess so, as the hydrogens can be/are anyway removed, but just to make sure.
So in case of tautomers, after removing the Hs the only difference left between the corresponding MCS parts would be the distribution of single and double bonds between the atoms. Should not matter or do you think this could create any foreseeable trouble?
To be honest, the answer is “I dont know”! I it depends on whether the RDKit substructure matcher matches the two sets of atoms or not. There is tautomer standardization in RDKit (the MolVS code of my colleague Matt Swain was ported to RDKit as a Google Summer of Code project a couple of years back, and I know there are Java wrappers), but I dont know if it has made it to KNIME (yet!) - I guess pre-applying that should make sure it works!
I have used this nice node combination that Steve created and it works nicely for the original problem.
However I tried to adapt it to my current problem, but did not succeed, so I will put a follow-up question here, (hope this is the right way, instead of a new thread … bit is directly related to the customized nodes provided here, … @ScottF, if you think this should anyway rather be a new thread, we can move it or I repost as new topic or so)
My Problem:
How could I use this node combination to compare a whole list of molecules (chemically identical , but different 3D positions of the atoms), in a way to filter out the 3D poses which are very similar to each other. (i.e. exploiting the nice 3D comparison build-in into the snippet already)
Example:
Molecule 1 vs Molecule 2 → RMS 1.5
Molecule 1 vs Molecule 3 → RMS 0.8
Molecule 1 vs Molecule 4 → RMS 1.5
Molecule 1 vs Molecule 5 → RMS 0.1
…
Molecule 2 vs Molecule 3 → RMS 0.7
Molecule 2 vs Molecule 4 → RMS 0.0
…
Molecule 3 vs Molecule 4 → RMS 1.1
Molecule 4 vs Molecule 5 → RMS 1.4
…
Molecule 2 vs Molecule 4 → RMS 0.0
…
If a cutoff of RMS = 0.2 is applied:
→ Molecule pairs 1 and 5 & molecules 2 and 4 are then too similar, respectively,
so final list should only contain:
Molecule 1
Molecule 2
Molecule 3
How can I do this … it should be possible with these nodes and some clever loops. I tried quite a bit, but could not come up with a working solution.
Remove all the rows where the left row and right row are the same (these are going to give - hopefully! - a distance of 0.0, and confuse things!)
Calculate the RMSD between the pairs
Remove any row with a distance greater than your threshold
Use Duplicate row filtering
Alternatively (and this is how the RDKit RMSD prune works) you need a recursive loop with 2 inputs. Into the first input, put your first molecule (this is going to be your ‘keeps’ table), and put the rest of the table into the second input. Split the 2nd table so that you have the first row (next ‘test’ molecule) and ‘the rest’, and cross join the ‘test’ with the ‘keeps’. Calculate the RMSD and then use groupby to find the minimum distance - if it is > the threshold, then add the ‘test’ row to the ‘keeps’ table. Now send the ‘keeps’ table and ‘the rest’ back around the recursive loop until there are no more rows left to test.
thanks again for the great ideas. I fiddled around with that but could not get it to work. My main problem with the crossjoining that your solution (post 10) requires 2 inputs to compare with each other. So I would need to crossjoin and then split again for the calculation, but how to store the result when re-joining … ideally in some kind of matrix or ideally in a way, that one can feed it in the Distance Matrix Calculator Node in order to perform a hierarchical clustering (with the respective Node for that).
Would you have an idea how to link this, i.e. clustering the poses by their RMSD distances?
I feel like both parts (RMSD comparison, see your solution) and clustering nodes are already there.
Or would a connection be much more difficult?
 ), but when I then want to look at the output of the cross-joiner or the Java snippet node, then my GUI crashes
), but when I then want to look at the output of the cross-joiner or the Java snippet node, then my GUI crashes  … probably something specific to my system, so I opened a ticked with the support. As soon as this is resolved, I will try it out more.
… probably something specific to my system, so I opened a ticked with the support. As soon as this is resolved, I will try it out more.