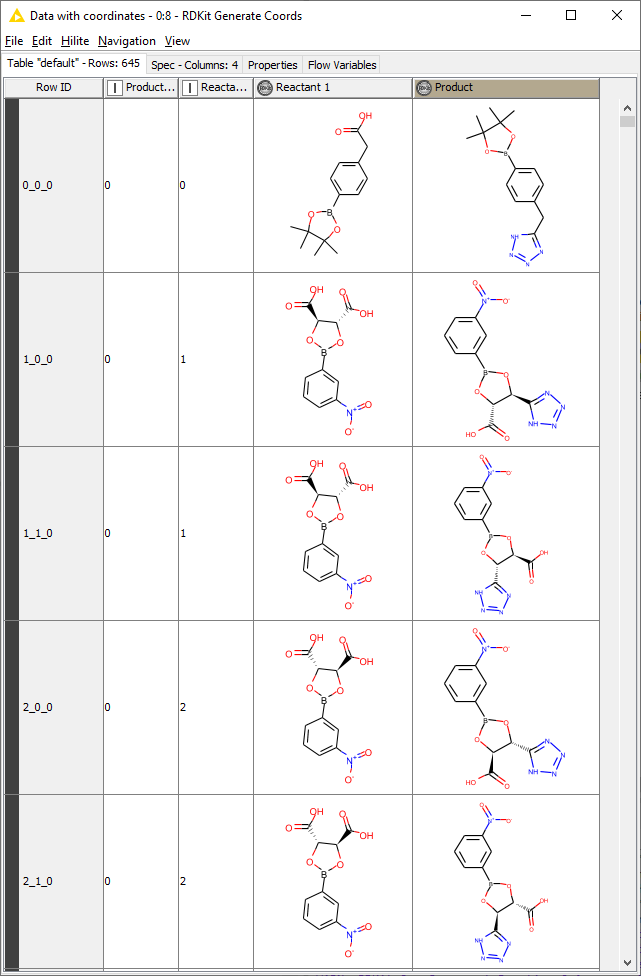
Hi fine people,
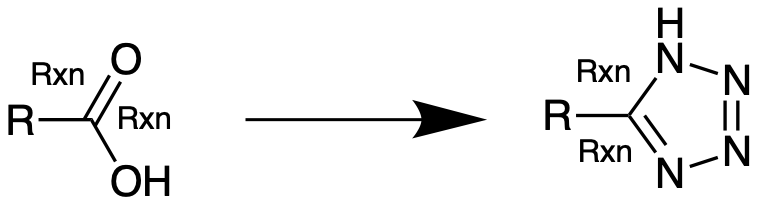
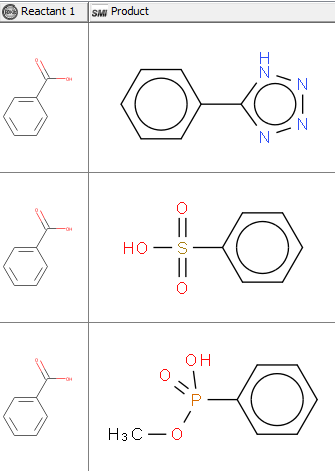
I am trying to create a KNIME workflow that would accept a list of compounds and carry out bioisosteric replacements (we will use the following example here: carboxylic acid to tetrazole) automatically.
NOTE: I am using the following workflow as inspiration : RDKit-bioisosteres (myexperiment.org). This uses a text file as SMARTS input. I cannot seem to replicate the SMARTS format used here.
For this, I plan to use the Rdkit One Component Reaction node which uses a set of compounds to carry out the reaction on as input and a SMARTS string that defines the reaction.
My issue is the generation of a working SMARTS string describing the reaction.
I would like to input two SDF files (or another format, not particularly attached to SDF): one with the group to replace (carboxylic acid) and one with the list of possible bioisosteric replacements (tetrazole). I would then combine these two in KNIME and generate a SMARTS string for the reaction to then be used in the Rdkit One Component Reaction node.
NOTE: The input SDF files have the structures written with an
attachment point (*COOH for the carboxylic acid for example) which
defines where the group to replace is attached. I suspect this is the
cause of many of the issues I am experiencing.
So far, I can easily generate the reactions in RXN format using the Reaction Builder node from the Indigo node package. However, converting this reaction into a SMARTS string that is accepted by the Rdkit One Component Reaction node has proven tricky.
What I have tried so far:
-
Converting RXN to SMARTS (Molecule Type Cast node) : gives the following error code :
scanner: BufferScanner::read() error -
Converting the Source and Target molecules into SMARTS (Molecule Type Cast node) : gives the following error code :
SMILES loader: unrecognised lowercase symbol: y- showing this as a string in KNIME shows that the conversion is not carried out and the string is of SDF format :
*filename*.sdf 0 0 0 0 0 0 0 V3000M V30 BEGINetc.
- showing this as a string in KNIME shows that the conversion is not carried out and the string is of SDF format :
-
Converting the Source and Target molecules into RDkit first (RDkit from Molecule node) then from RDkit into SMARTS (RDkit to Molecule node, SMARTS option). This outputs the following SMARTS strings:
- Carboxylic acid :
[#6](-[#8])=[#8] - Tetrazole :
[#6]1:[#7H]:[#7]:[#7]:[#7]:1
- Carboxylic acid :
This is as close as I’ve managed to get. I can then join these two smarts strings with >> in between (output: [#6](-[#8])=[#8]>>[#6]1:[#7H]:[#7]:[#7]:[#7]:1) to create a SMARTS reaction string but this is not accepted as an input for the Rdkit One Component Reaction node.
Error message in KNIME console :
ERROR RDKit One Component Reaction 0:40 Creation of Reaction from SMARTS value failed: null
WARN RDKit One Component Reaction 0:40 Invalid Reaction SMARTS: missing
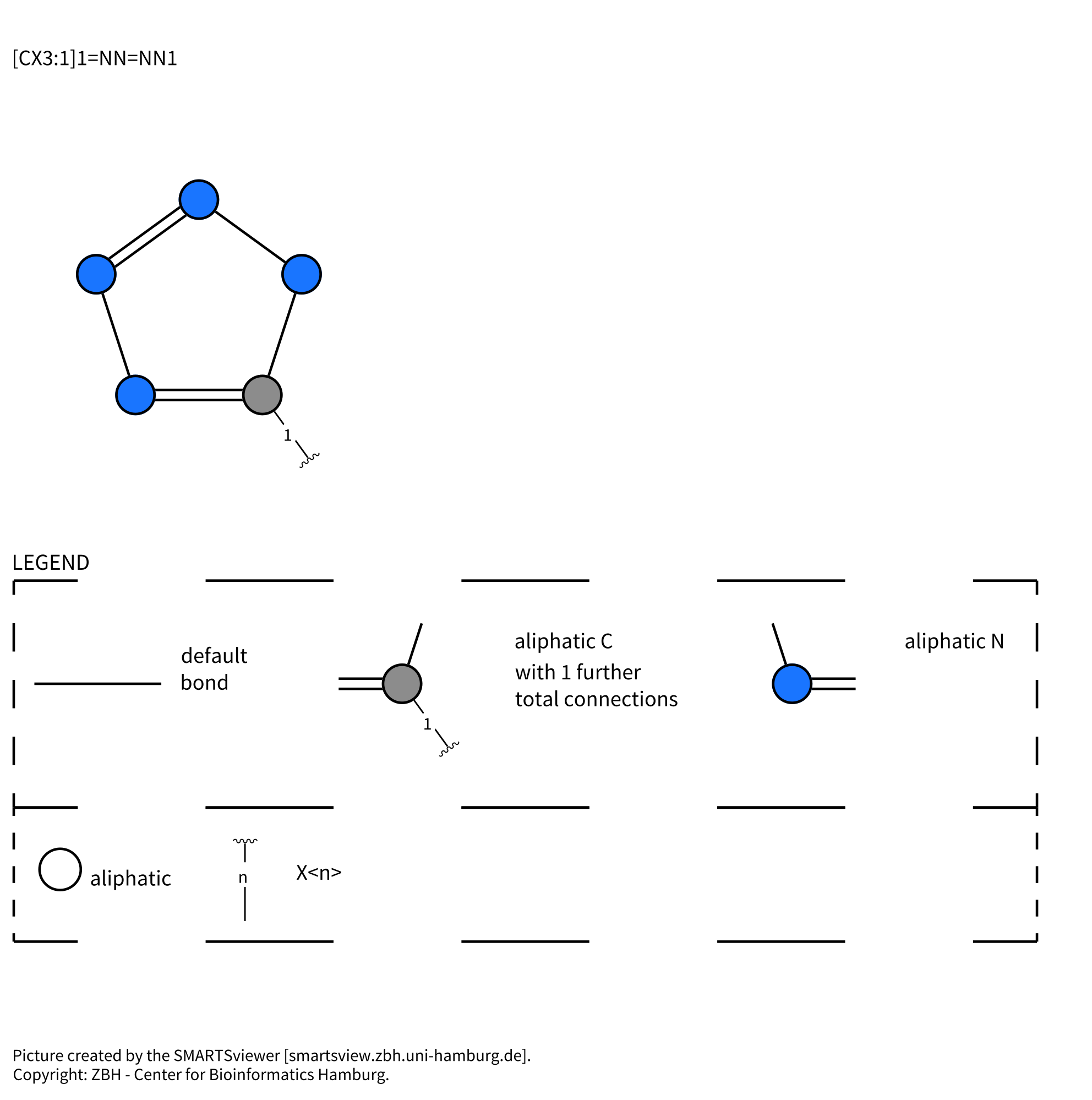
Note that the SMARTS strings that this last option (3.) generates are very different than the ones used in the myexperiments.org example ([*:1][C:2]([OH])=O>>[*:1][C:2]1=NNN=N1). I also seem to have lost the attachment point information through these conversions which are likely to cause issues in the rest of the workflow.
Therefore I am looking for a way to generate the SMARTS strings used in the myexperiments.org example on my own sets of substituents. Obviously doing this by hand is not an option. I would also like this workflow to use only the open-source nodes available in KNIME and not proprietary nodes (Schrodinger etc.).
Hopefully, someone can help me out with this. If you need my current workflow I am happy to upload that with the source files if required.
Thanks in advance for your help,
Stay safe and healthy!
-Antoine