For my Bachelor’s thesis I’m using KNIME and try to analyze molecules and classify them.
I have to questions and would be grateful if you could help me.
I wanted to count the atoms (C, N, O) from a SMILES but all nodes I tried could not help me. Do you have a solution and the right node for me?
Also I want to know if there’s a possibility to know which functional group my molecules have.
Which atoms exactly do you want to count? What have you tried? If you just want C, N, and O in SMILES strings, then the Speedy SMILES Element Count (C,N,O) node will work.
If you want to count other atoms then you’ll need to create SMARTS queries for the atoms in question and use either the SMARTS Query node or the RDKit Substructure Counter do the counting. This is how you’d count specific functional groups as well.
Hi @elsamuel
Thank you for your fast answer!
The “Speedy SMILES” I’ve tried but and I had a table with SMILES entrys but if I want to configure the node there pops up a window with following text: “Dialog cannot be openend - The dialog cannot be openend for the following reason: No column in spec compartible to ‘SmilesValue’”.
Since I’m not used to SMARTS I need to try the other nodes.
In that case you need to convert the column with smiles values. When you read from a text file or excel file with file read, the column type is then string by default. You then need to use the molecule type cast node to convert the column to a molecule type. Then it will work.
Thank you for your help. I was possible to count the elements.
But I was not able to get a list of my functional groups. I’m not sure which two inputs the node needs. On the one hand I have the molecule structures and then I need a list with the functional groups. Maybe you have a tip for me?
Okay, I will try to share it and explain the problem. I’m a chemist and maybe some things are for you easier and quit simple to solve… Sorry for that.
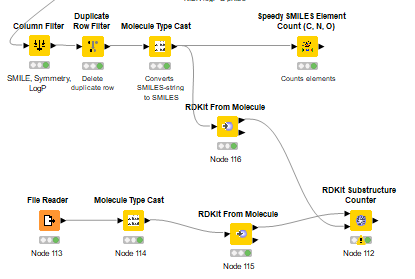
This is my workflow at the moment. The File Reader has a CSV with funcitonal groups as SMILES. Then I try to convert the strings so the RDKit could use it. At the moment I have a table at the RDKit Substructure counter but every entry is 0.
I’m pretty sure you need to connect the RDKit Substructure Counter node differently. Eg queries (=functional groups) go to the lower port and the molecules to the upper port.
It is like bewitched… I tried it the whole day in the different way and it did not work. Finally I have my table with the right entries… That’s embarrassing. Anyway, it’s Friday and I finally have a solution for the problem. Thanks to all of you for your help!
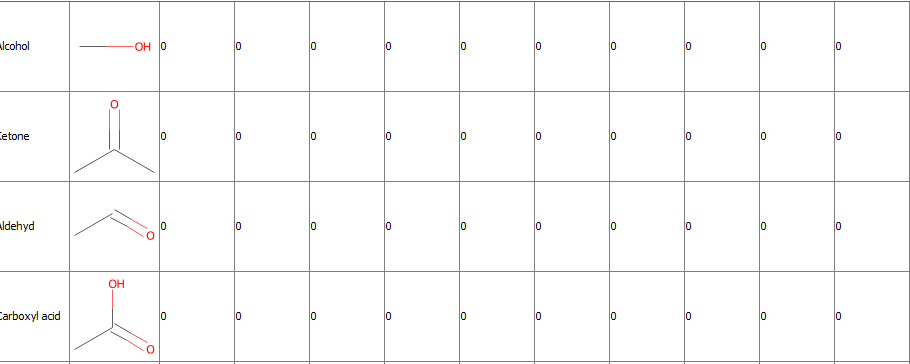
First of all thank you again for your help. The node counts my structures but not correctly. For example: I have a structure with an ether and a peptide bond. Unfortunately it counts 2 alcohol, 1 aldehyd, 2 amines …
I tried other nodes to solve it but nothing works and I always have errors.
Do you have an idea?
Cheers!
If you use the substructure counter, it depends on your definition of the query if it works or not but in this case I think there is a setting about only matching once because depending on your pattern definition, the same alcohol can match twice.
This setting you mentioned I tried but it did not work either. I also tried the other node “SMARTS Query”. I have a result but its not clear which atoms are counted. Maybe I need to try an other way with or without KNIME.
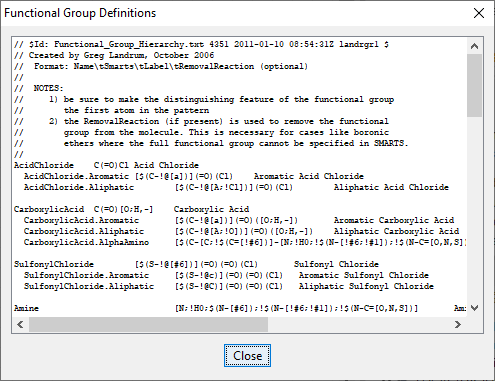
I think the problem is that a lot of your functional group definitions are not appropriate.
All of your queries need to be overhauled, and they need to be in a SMARTS format.
For example,
you define an ether as COC. This is going to match aliphatic esters.
you define an amide as CC(=O)N(C)C. This is only going to match specific amides with 2 aliphtic carbons on the nitrogen and another aliphatic carbon on the other end
you define an amine as CC(N)=O. That looks like a generic amide
you define a nitrile as C=N. That’s a double bond, while a nitrile has a triple bond.
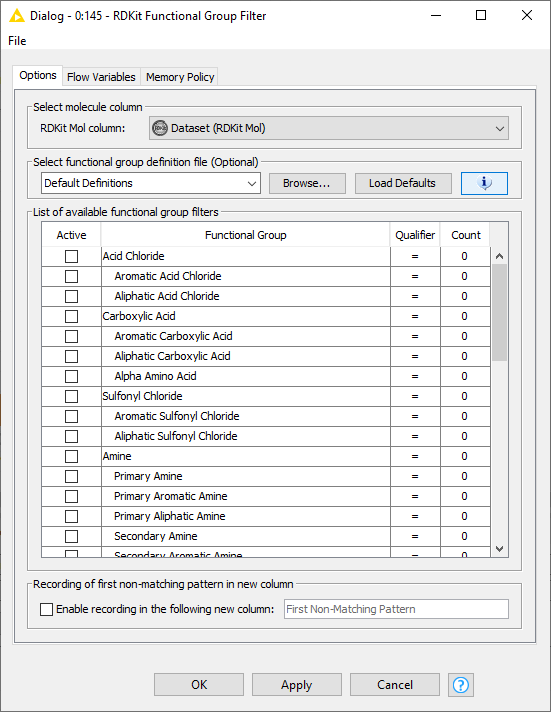
You can find SMARTS examples in the RDKit Functional Group Filter node. Just click the i icon and you’ll get the default list:
You can also find them on the Daylight website, or in papers like this one and this (check the supporting information), or you can use this SMARTS editor.