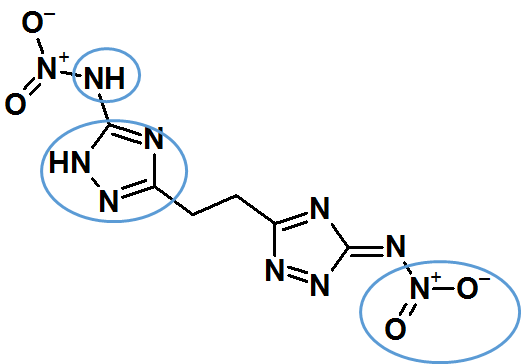
I’m trying to create 3D structures with the Generate Coordinates node from a library of ~2m compounds, however around 200,000 of them fail, giving me the error ‘MolSanitizeException’. After going through the failed compounds and trying some custom compounds, I think the problem is compounds with charges. . These charges can’t be removed by simply adding or removing hydrogens. Is there a way to ignore this error and still generate 3D structures with the charges?
I can see several reasons why this specific structure might fail.
Treating nitrogen atoms and the successive addition of protons correctly is fairly difficult for most softwares.
Within Knime it also depends which formats you start with/convert towards. Are you starting from SMILES codes? If so, from my experience there’s a few possibilities why they might fail:
naked Pyrrole structures:
If you have a Pyrrole or related structure (here the diazapyrrolic subunit) I’ve learned that it’s important to specify the proton within the square brackets: c1ncn[NH]1
Amine substituents (here your aliphatic NH group):
I’ve encountered problems with these as substituents on aromatic systems as well. The logic within RDkit doesn’t always translate them well, drops the protons and gives a wrong structure. I found that an extra step of correcting the SMILES code (either by hand or by string replace) helped take care of that. So, even when you manage to generate 3D structures it might be prudent to check the output again.
String replacement within SMILES can be rather messy when you do it for a batch of molecules and it requires some thought and experimenting - but it can work.
nitro groups:
I don’t have a whole lot of experience with NO2. But if you draw nitrobenzene with explicit charges and feed it into the following workflow:
Marvin Sketch (output to sdf cell) => RDKit from Molecule => RDKit Canon SMILES
it generates this SMILES code:
O=[N+]([O-])c1ccccc1 (works)
Have a look whether your compounds meet this format. If they look more like this
O=N(=O)c1ccccc1 (doesn’t work)
then RDKit doesn’t seem to parse them.
So, in conclusion:
a) I’d break the molecule down to all possibly problematic substructures
b) check RDKIT to see if it has problem converting these
c) check the input files / strings for possible ‘inconsistencies’
d) potentially change the SMILES code - manually and/or by string replacement.
 . These charges can’t be removed by simply adding or removing hydrogens. Is there a way to ignore this error and still generate 3D structures with the charges?
. These charges can’t be removed by simply adding or removing hydrogens. Is there a way to ignore this error and still generate 3D structures with the charges?