Hi all,
Are there any working Knime examples of deep learning applications (DeepLearning4J, Keras, TensorFlow) where chemical structures are used as input to train models for biological activities etc.
Cheers/Evert
Hi all,
Are there any working Knime examples of deep learning applications (DeepLearning4J, Keras, TensorFlow) where chemical structures are used as input to train models for biological activities etc.
Cheers/Evert
Hi Evert!
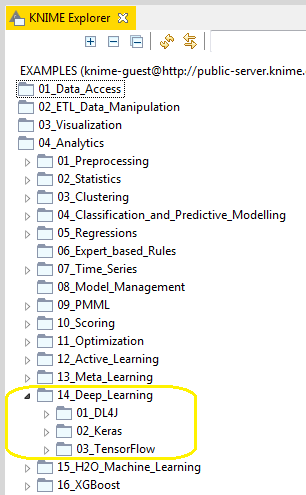
Have you checked workflows from KNIME Examples server? Under 04_Analytics --> 14_Deep_Learning you have examples workflows for above mentioned applications…
Br,
Ivan
Hi,
None of these examples use chemical structures as input, I believe that is the bottleneck: how to describe chemical structures in a format that deep learning networks can understand.
Thanks/Evert
Hi Evert,
thanks for you question. Actually we don’t have any published examples on deep learning using chemical structures but we are working on it.
Let me ask you first whats the data format you are using for chemical data?
In general it doesn’t matter if it’s a SDF, CSV or something else. In this data set you should have at least a chemical structure like SMILES for example and a column with the category you want to predict (like activity).
If you have your own descriptors that’s also okay - but you also can calculate descriptors in KNIME using the RDKit Descriptor Calculation Node for example.
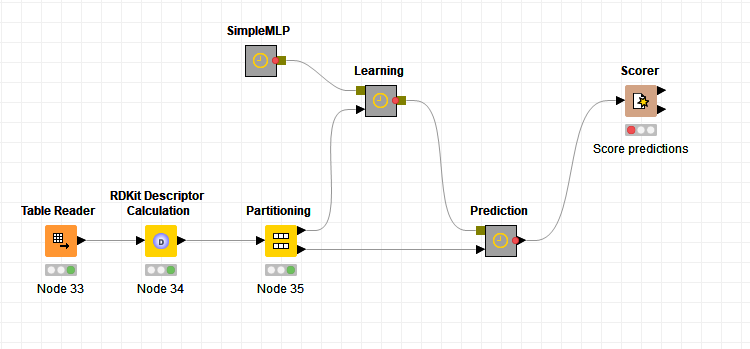
If you want to try an example you could merge two workflows from the KNIME Examples Server.
For the data you could take the one used in 08_Model_Optimization_and_Selection and then
replace it in 03_Network_Example_Of_A_Simple_MLP, calculate descriptors and then do configuration on the learning model.
Could look for example like this then:
Best,
Martyna
Hey Martyna,
Thanks for your suggestions. I have indeed a large data set of structures in Smiles format, and was thinking that I could generate structural fingerprints (e.g. Morgan with RDKit) and then expand them with the Fingerprints Expander node from the Erlwood contributions. This would give 1024 columns describing each structure as input, together with the activity class.
Will have a shot at it and let you know. Am also looking forward to what Knime has in the pipeline in this context.
Cheers/Evert
Hi Evert,
does fingerprints give you any information about the molecular properties of a chemical sturcture?
For my understanding it is useful to extend your data with structural descriptor or fragmentation informations.
Best
Hermann
It depends on the problem at hand. Some parameters (e.g. solubility) are strongly driven by logP, whereas others can be more dependent on the presence of specific substructures (e.g. toxicity). They can of course also be combined.
Best/Evert
My opinion on that would be that it’s a waste of time. Just feed the descriptors and/or fingerprints into Random Forest or xgboost. That will usually work better and easier than deep learning.
If you want to look into deeplearning, I’m afraid you will need to leave the knime environment mostly. The strength of deeplearning is the generation of it’s own features and not using preexisting features. The key word here is graph convolution neural network and in application it’s deepchem.
In my experience RandomForest and XGBoost often overfit, i.e. they work very well on training sets but not on test sets.
At the risk of stating the obvious: you can tune the hyper-parameters of ensembles of trees (both random forests and GBMs) to minimise overfitting and get the best performance on the validation/testing sets. Tree depth, sampling fraction and the number of trees would be the first candidates for optimisation
Hi,
At the risk of asking the obvious, which of the XGBoost Tree Ensemble Learner setting correspond to the variables you suggest to optimize?
Kind regards,
Evert
As long as the predictions are “good enough” especially doing cross-validation then the overfitting isn’t really a problem. Cross-validation is important to see how the performance depends on the training set. If you have big variance between different folds, then yes the overfitting is a problem, if not not really big of an issue.
And keep in mind that deep neural networks are much, much more prone to overfitting and harder to tune. Fear of overfitting is an argument against deeplearning.
In if we are talking about overfitting and cross-validation,this publication is also highly relevant. (paywalled)
Tree Ensemble / RF in knime is pretty stable and not much affected by params. Maybe important are Fraction of samples per tree (0.7 usually works well) and setting the sampling to stratified especially if classes are imbalanced. Also obviously limiting tree depth directly or indirectly can limit overfitting.
xgboost is much more affected by the params. IMHO the default learning rate (eta) is way too high but this has nothing to do with overfitting. In case classes are unbalanced, scale positive weight is important. Lambda = L2, alpha = l1, well in fact it’s explained in the node description or xgboost docs…So I’m just repeating the obvious here.
Personally I always start with RF because that tells you quickly if there is some signal in the data set. You have to be aware that deep learning mostly “only” add a small edge on top of RF/xgboost and it’s not always worth the orders of magnitudes more computation needed. Don’t buy too much into the hype.
Let me add a bit to the nice answer by @beginner.
One can find also nice intro in cross validation also free of charge. Google gives many results, for example this one is very illustrative: https://towardsdatascience.com/cross-validation-70289113a072
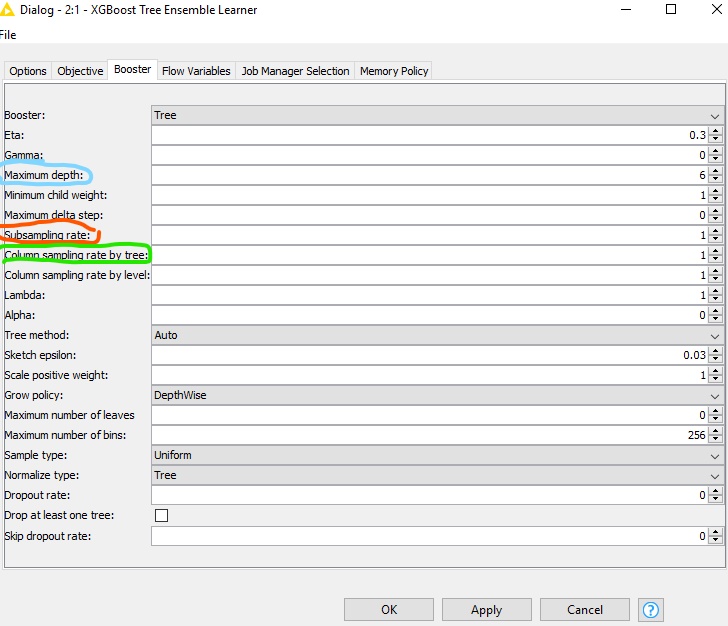
Regarding the model parameters, you seem to use XGBoost GBM model. I have highlighter the tree depth in blue and sampling fractions (row and column sampling) in red and green.
Boosting rounds) in the first tab. The rule of thumb for good starting values is sampling fractions =~ 0.7, tree depth =~ 5..15. The number of trees depends on the dataset and will have to be optimised. Note that large tree depth or number of trees can lead to overfitting and the definition of “large” depends on the dataset
And just for completeness sake: This applies for gradient boosting.
Tree depth also affects overfitting in Random Forest. however adding more trees in Random forest does not increase overfitting!!
This topic was automatically closed 182 days after the last reply. New replies are no longer allowed.