I am trying to exploit SDF structure on a “flagged library”. This library is composed of many scaffolds having 2 Iodines “flagging” the exit vector positions.
I have 2 tasks on this library:
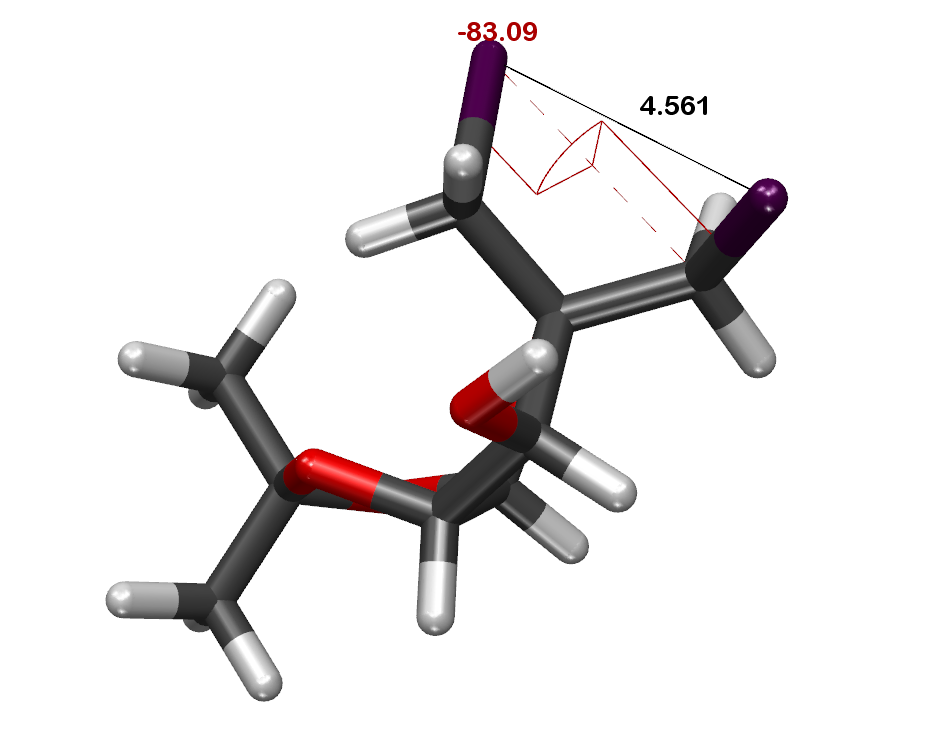
1) Get the distance between the 2 Iodine atoms.
2) Get the torsion angle between the 2 exit vectors.
I have managed to extract the coordinates of my 2 Iodines atoms to get the distance with a not very elegant succession of string manipulation nodes. So “1)” is done.
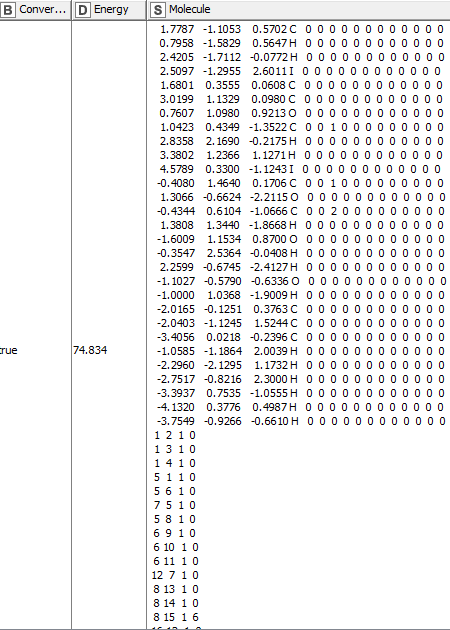
Regarding 2), I would really appreciate some help. Each molecule has the following structure: a first block (here in bold) of atomic coordinates and the second block of bond description. The first block has X Y Z coordinates and then the atomic element followed by characterisations of that element that I do not need for that applications (mostly 0s and somes 1s). The second block is composed of 4 digits of which only the first 2 are important. They describe which atoms are linked together by their numbering which is determined by their row number in the first block).
I need to:
=> numbering the row inside that string cell
=> identify the row in which the 2 iodines are located (row 4 and 27 in the example below)
=> go to the bond description block and find, within the first 2 digits of each row, either 4 or 27
=> Extract the other “atom number” of each pair number (here 1 and 22, in bold as well)
=> Use these 2 new numbers to extract the coordinates of the corresponding atoms in the atom block
The output should be essentially 12 new columns, Iodine 1 X, Iodine 1 Y, Iodine 1 Z, Partner 1 X, Partner 1 Y, Partner 1 Z, Iodine 2 X, …, Partner 2 Z.
I would really appreciate any help! Also if someone knows how to match for any letter in the sting manipulation node that would also allow me to finish the project, separating the atom block from the bond block by doing a backward search for the last letter and then getting the “row numbers” with a \n cell splitter.
Hi @avalery can you elaborate more on those lines you wrote?
Also, I have 2 additional queries:
The two blocks; are they inside the same table in KNIME? Or do they belong to different tables? Perhaps you can upload the dataset.
You mentioned ‘numbering the row inside that string cell’. Not sure which cell you’re referring to, which brings me back to my first question about how the dataset is structured in a tabular form.
Sure. Essentially you have 4 atoms involved in the 2 exit vectors. In this example it is one C-I bond and one N-I bond.
This means that you have 4 “lines of interest” in the atom block. Here, the lines that interest us are 1 and 4 as the first pair and 22 and 27 as the second pair.
1&4 -3.2355 0.3926 -0.4140 C 0 0 0 0 0 0 0 0 0 0 0 0 -3.2674 2.2798 0.5824 I 0 0 0 0 0 0 0 0 0 0 0 0
searching for the lines containing “I” (letter i in capital). Here 4 and 27
Figuring out to which atoms they are connected. Here 1 and 22 (these are the 2 “new numbers” I was referring to)
Once you have figured out which atom are connected to Iodine, you need to create the 2 vectors based on the two pairs of atoms.
So, with Partner 1 being on line 1 of the atom block, and Iodine 1 being on line 4 of the atom block and Partner 2 being on line 22 and Iodine 2 being on line 27:
Vector 1 = sqrt((Iodine 1 X - Partner 1 X)^2+(Iodine 1 Y - Partner 1 Y)^2+(Iodine 1 Z - Partner 1 Z)^2)
Vector 2 = sqrt((Iodine 2 X - Partner 2 X)^2+(Iodine 2 Y - Partner 2 Y)^2+(Iodine 2 Z - Partner 2 Z)^2)
Yes, they are even in one single cell, making the “row numbering” part a hassle.
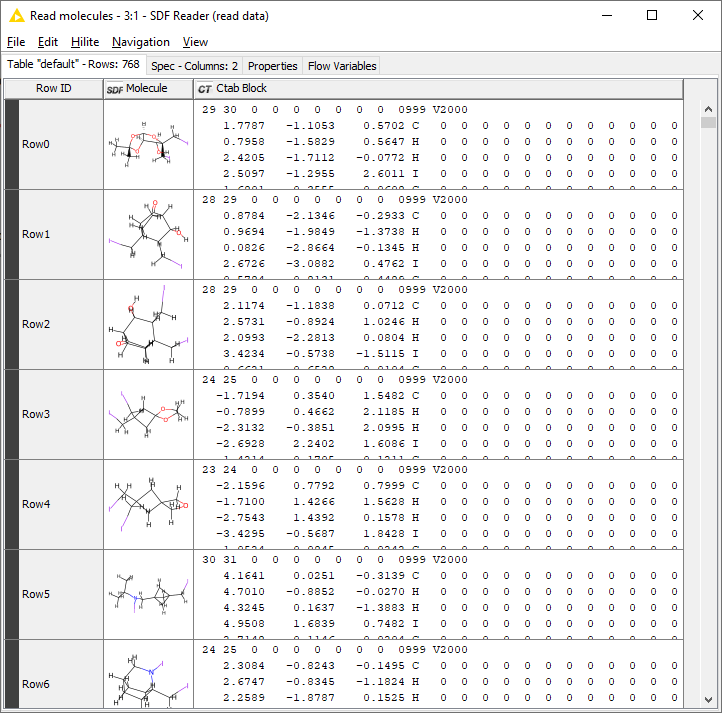
Hi @avalery I’ve tried using an SDF file from one of the workflows in the hub, but it seems like your bond block has a different format. Here’s what I have (after converting it to a workable table):
Because of the different formats, it wouldn’t matter what I do since my workflow won’t be applicable to your dataset.
Unless you upload your dataset (or even better, the workflow itself), I can do nothing further.
My SDF are generated with RDKit, I guess that can explain the different format. I am sorry but I am not allowed to share the workflow. I cannot attach SDF as it is not authorized by the website. I saved it as text if you would like to take a look.
Unfortunately a .txt file will appear differently when being read in KNIME. It’s a totally different thing. I’m afraid this is where we diverge our paths. Wish you all the best & hope you found the solution!
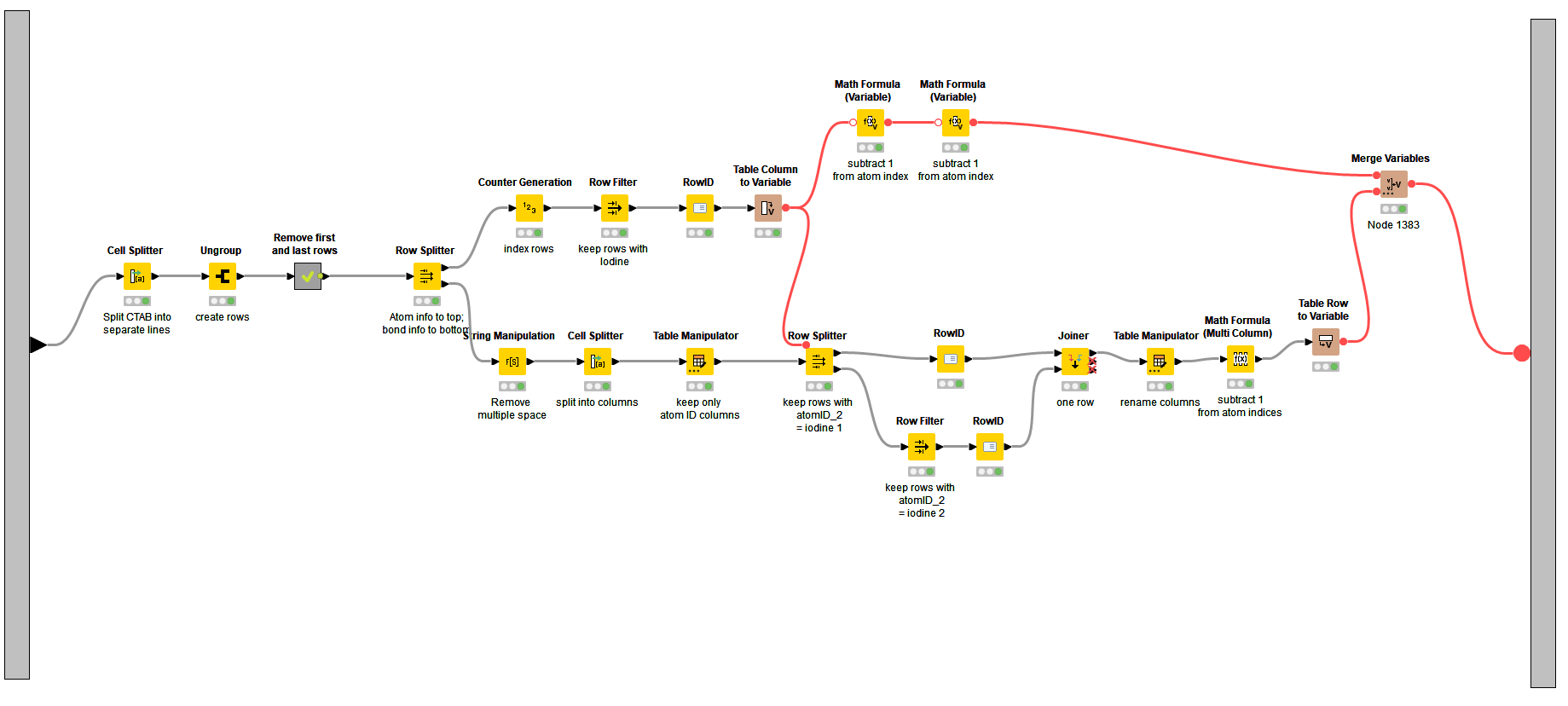
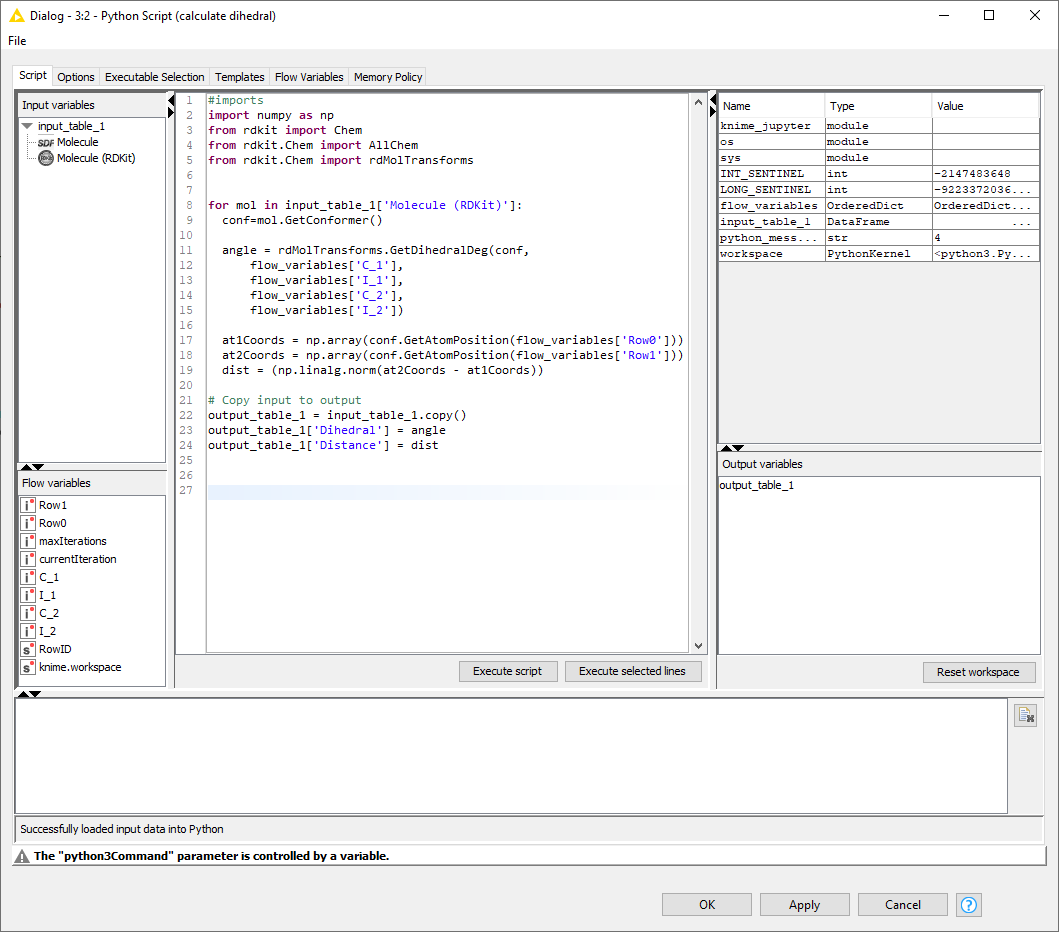
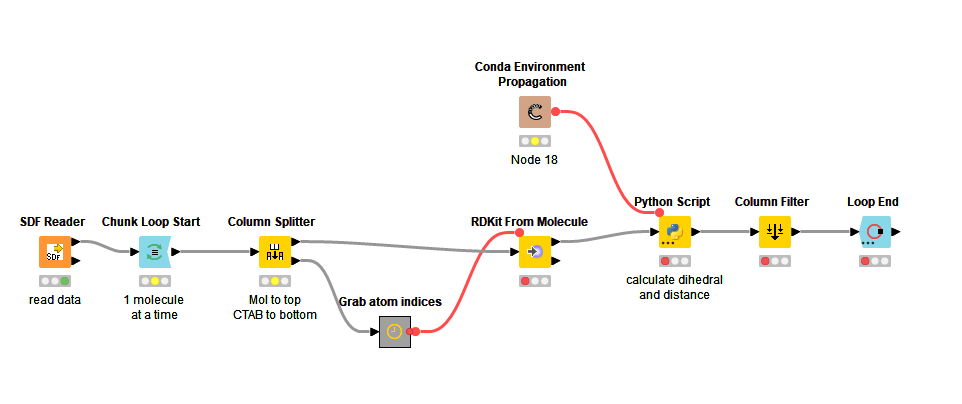
Then working on each molecule individually, I parse the CTAB block to find the atom indices of the iodine atoms and the connected carbon atoms, and convert the atom indices to flow variables:
Thank you so much! I will try to use it and set up anaconda!
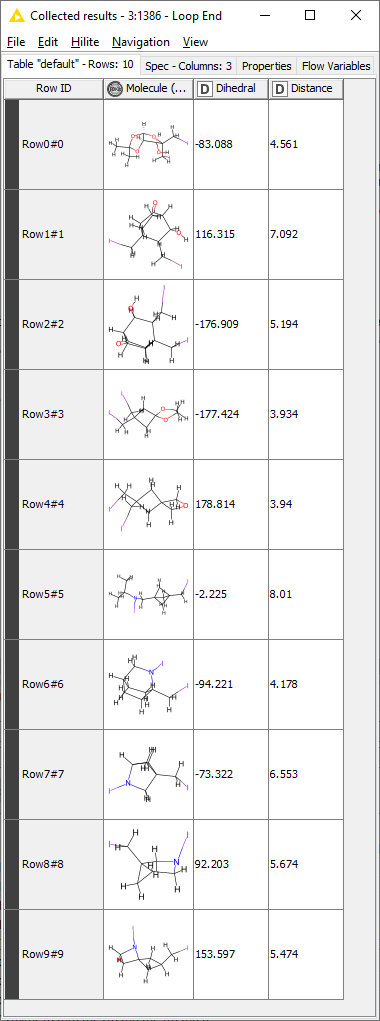
Yesterday I managed to get to isolating coordinates for all the involved atoms but I am using so many Nodes … This solution seems so much more elegant!