I got some data from single-cell transcriptomics in an h5 file. I want to have a look at the raw data and analyze it in KNIME. However, I am struggling to extract the data from the file and place it in a table in KNIME. Does anyone know how to do this? Perhaps some examples to follow will be amazing.
Thanks for the idea. My problem is that I don’t know the code to export this document to the table format. That is why I was asking for some sort of example.
Hi @VAGR_ISK , I’ve never worked with H5 files before, but looking at old form threads, somehow this node is involved. Not sure if that applies to your case. (I’ve never worked with this node too so I don’t know anything about it).
Other than that, from a little bit of Googling, there are ways to convert H5 files to CSV. Maybe if you can find a way to do it, then you may read the CSV file in KNIME.
Details for the h5py package, with examples on reading hdf5 files is here.
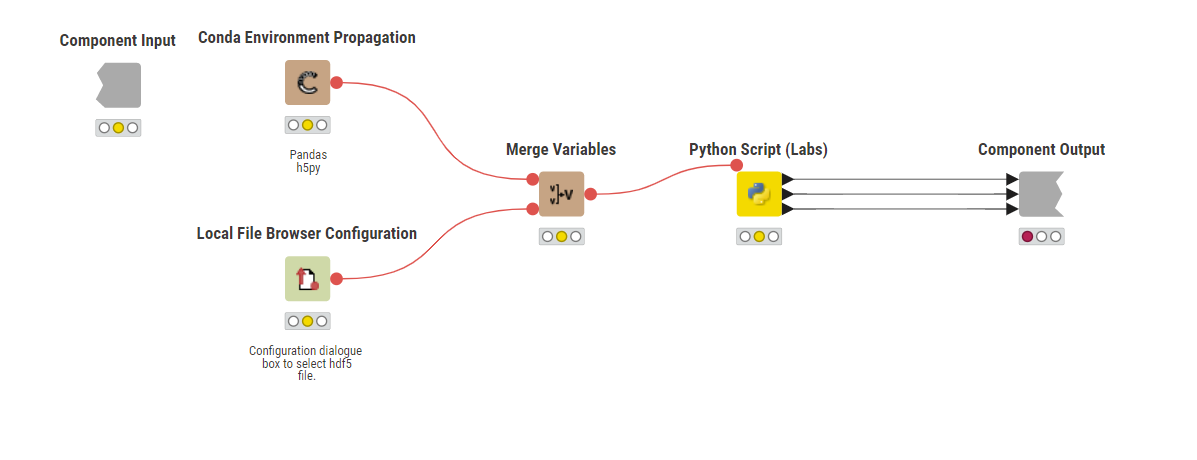
The Python code is as follows, it should be well enough commented to follow what is going on. The hardest part is getting the data types stored in the hdf5 file into a data type that KNIME understands - in both cases I converted unsigned-ints and bytes to strings.
The code opens the file based upon a variable input (passed from the configuration node). It then opens the file and creates Pandas dataframes using a dictionary. The dictionary keys are column headings in the table, the rows come from the numpy tables (which in this case are all single column series) in the file.
Once the value types have been changed the dataframes are written to the three outputs of the Python node.
import h5py
import knime_io as knio
import pandas as pd
# Get the file to read from the flow variable.
file_to_read = knio.flow_variables['file-input']
# Open the h5py as a file object
with h5py.File(file_to_read, "r") as file_object:
# Create the output table as a Pandas dataframe.
# Use Python dictionary to structure dataframe.
output_table = pd.DataFrame({
"barcode": file_object["barcode"],
"barcode_corrected_reads": file_object["barcode_corrected_reads"],
"conf_mapped_uniq_read_pos": file_object["conf_mapped_uniq_read_pos"],
"gem_group": file_object["gem_group"],
"genome": file_object["genome"],
"nonconf_mapped_reads": file_object["nonconf_mapped_reads"],
"reads": file_object["reads"],
"umi": file_object["umi"],
"umi_corrected_reads": file_object["umi_corrected_reads"],
"unmapped_reads": file_object["unmapped_reads"],
})
# Note: KNIME cannot use uint64 or uint32, so convert to string
output_table = output_table.astype(str)
output_genes = pd.DataFrame({
"gene_ids": file_object["gene_ids"],
"gene_names": file_object["gene_names"],
})
# Note: KNIME doesn't support binary type, therefore decode to string
# assume utf-8 encoding.
output_genes["gene_ids"] = output_genes["gene_ids"].apply(
lambda el: el.decode("utf-8")
)
output_genes["gene_names"] = output_genes["gene_names"].apply(
lambda el: el.decode("utf-8")
)
output_genome_ids = pd.DataFrame({
"genome_ids": file_object["genome_ids"],
})
output_genome_ids["genome_ids"] = output_genome_ids["genome_ids"].apply(
lambda el: el.decode("utf-8")
)
# Write the Pandas dataframes to output ports
knio.output_tables[0] = knio.write_table(output_table)
knio.output_tables[1] = knio.write_table(output_genes)
knio.output_tables[2] = knio.write_table(output_genome_ids)
@VAGR_ISK there are several threads out there on Stackoverflow about a genuine approach to extracting data from h5 files but right now I was not able to make anything work. Would still be worth exploring I think.
Thanks for that great workflow. I could replicate exactly what you did. However, I am actually working with the matrix data file, which is the other file that appears in the link from where you got the sample data in the component that you sent me (GSM4735548_H18_19086_TB.raw_gene_bc_matrices_h5.h5). If I try to use the matrix file in your phyton script node, python tells me that it was expecting an intended block.
I managed to read the matrix file using scanpy see code below.
However, I am not able to place this in an output table, because KNIME only accepts Panda DataFrames as output. Would that be a way to convert the scanpys’ results in a Pandas’ DataFrame so that KNIME can arrange it in an output table?
Moreover, since this is a single cell transcriptome dataset, in the output I need the gene_id or gene_name (rows) linked to the expression of each gene in each cell (Biological cell, not table cell). Each cell will be in the columns. See the skim table screenshot below.
I am not really a coder. Therefore, it is difficult for me to find ways around it. Since I need coding for this. The h5 I am talking about is the output of a processing system that uses these matrices and I am not sure that they are making the things easy for us to extract the data out of these files since they want us to pay for the software. In my case, I don’t mind paying for that software but I am not getting what I want for analysing the results in KNIME which is as in the table below.
@mlauber71 actually there is not much I have done in KNIME aside from what I showed to @DiaAzul. However, I can share a Jupyter notebook with a file similar to mine that is from a publication (Here). I did not make this notebook, I just copy/pasted some of the code from a friend. Perhaps that can give you some ideas. However, the code in the Jupyter is not giving me the disposition of the data as need it (As I showed in the table above), it is only an example of how other people work with those single-cell transcriptomic matrices files to extract the data. It will be amazing to have a node or components that can handle single-cell transcriptomics like this in KNIME.
@VAGR_ISK, thanks for posting your problem. It is a good use case to identify challenges in using Python with KNIME.
The good news is that you can read the file into KNIME, the bad news is it is very slow (hours slow). The bottleneck is importing the pandas dataframe into a KNIME table - which is slow. I suspect this is because the table is 33,694 columns which, to the best of my knowledge is supportable by KNIME but is not efficient. A KNIME developer may have a better insight into this.
The scanpy library relies heavily on sparse matrices - this makes both storage and processing efficient. If you can use Python then I would recommend that. The sparse data in the file is approximately 3.3Mbytes, as a dense matrix this increases to ~95Gbytes. I don’t believe that KNIME supports sparse tables, but may want to consider it for the future. Especially if there are many people who want to process this type of data (I suspect it is a growing market).
The code that I created for the Python Node is below. I only tested it on two batch iterations due to the time it was taking to load the entire file; however, if you are prepared to wait then it should work. Note that once it completes opening tables to view them is incredibly slow.
Note: Due to the size of the array when converted from sparse to dense (so that it can be saved to KNIME) it was not possible to do this in one go on my computer (32Gb memory). So, there is an option to process the data in chunks. The size of the chunk (MAX_CHUNK), currently 1Gb, if you have more memory on your computer.
You may get a performance improvement by removing columns/rows with no data.
import knime_io as knio
import logging
import numpy as np
import pandas as pd
import scanpy as sc
from math import ceil
from scipy.sparse import csr_matrix
LOGGER = logging.getLogger(__name__)
# Set max size of a chunk to 10 Gbytes
GBYTE = 1024 ** 3
MAX_CHUNK = 1 * GBYTE
# Assume datacell is np.float32 = 4 bytes
CELL_SIZE = 4
# Get the file to read from the flow variable.
matrix_file = knio.flow_variables['file-input']
# Open the matrix file and reads data into an annotated matrix (anndata)
adata = sc.read_10x_h5(matrix_file)
# Ensure variable names are unique.
adata.var_names_make_unique()
# Logarithmise the data matrix
adata.raw = sc.pp.log1p(adata, copy=True)
# The dense table may exceed the memory available
# So split into batches.
# Create KNIME processed table
processed_table = knio.batch_write_table()
# Convert raw X data to a sparse row matrix for efficient slicing.
sparse_row_matrix = csr_matrix(adata.raw.X)
# Calculate rows per batch and number of batches
rows, columns = adata.shape
batch_size = int(MAX_CHUNK / (columns * CELL_SIZE))
batch_count = ceil(rows / batch_size)
LOGGER.warning(f"{batch_count=}, {batch_size=}")
for batch in range(batch_count):
LOGGER.warning(f"Start processing {batch=}")
batch_frame = pd.DataFrame.sparse.from_spmatrix(
sparse_row_matrix[batch * batch_size : (batch + 1) * batch_size],
index=sc.get.obs_df(
adata[batch * batch_size : (batch + 1) * batch_size]
).index.values.tolist(),
columns=sc.get.var_df(adata).index.values.tolist()
)
LOGGER.warning("Created batch frame.")
batch_output = batch_frame.sparse.to_dense().fillna(0).astype(float)
LOGGER.warning(f"Created batch output of size {batch_output.shape}.")
processed_table.append(batch_output)
LOGGER.warning(f"Processed output {batch=}")
LOGGER.warning("Processing complete.")
# Write the processed table to output port
knio.output_tables[0] = processed_table