I’m trying to use FASTA Sequence Extractor (downstream from Load Text Files, which gets a file name from String Input) to read a Uniprot database. It seems to be running fine, except it never finishes. The progress meter at the bottom of the (KNIME) node reaches 99% and then stays there indefinitely. It doesn’t produce an error or anything else. I definitely see CPU activity and RAM usage coming from KNIME but there is no indication that FASTA Sequence Extractor will ever finish. As I write this, it has been stalled at 99% for over 15 minutes in the latest run; it took less than 1 minute to get to 99%.
On a similar note, is SWISS-PROT the best option to select for a Uniprot database? I don’t see Uniprot as an option in here.
thank you!
Vernalis 1.17.3.v201808160901, KNIME 3.6.1 64bit Windows
I think what you are seeing is the KNIME framework writing the table to disk (I have seen this behaviour with other nodes too). Do you have a big table coming into the node, or a large number of rows in the output? One way you could test this would be to wrap this node, its predecessor and some downstream nodes in a wrapped metanode and running in streaming execution. This is partly a result of the node being a 1 input row -> many output row node (i.e. a single FASTA cell can contain multiple sequences, which are each output on separate rows). The result of this is that the whole table has to be re-written to disk by the node, rather than just any new columns. In streaming mode this doesnt happen.
Regards the header, I think SWISS-PROT should be good for Uniprot. The header option only tries to add a little extra parsing of the header row to extract useful content, so if it doesnt work and you are interested in anything in the header, then select the ‘Other’ option, and check the ‘Extract full header row(s) from FASTA’ option, and process it using additional nodes. (If you are able to send a sample Uniprot header then that would be even better, and we may be able to add Uniprot to the list - this morning I cannot access Uniprot unfortunately to check this myself).
Thank you for your reply. In the case of the database I have most recently tried to import, I have 42,356 sequences and 468,953 total lines in the FASTA file. By my experience this is on the large side for a UNIPROT database but not outlandishly so. I did just try radically reducing the file size (by way of ‘head -50’) to see what would happen with the same flow and this worked so indeed the file size does seem to have an impact here.
Could you provide an example of the streaming workflow you describe? I’m not sure exactly how to construct what you are recommending here.
Take a look at https://www.knime.com/blog/streaming-data-in-knime - the basics is that you take your workflow, select the FASTA node and some nodes upstream and downstream from it, and right click -> ‘Encapsulate into wrapped metanode’. Then double-click on the wrapped metanode, which should look something like this:
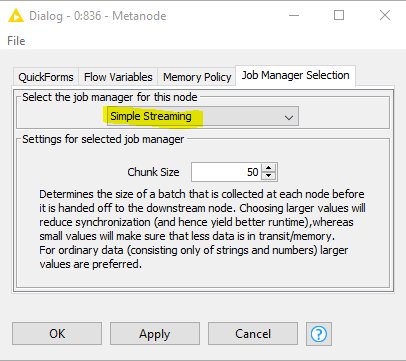
and in the Job Manager Selection tab of the resulting dialog, select Simple Streaming:
The above link gives more detail. You may need to also do some configuration inside the wrapped metanode if you have flow variables involved at all. To do that, ctrl+double-click on the metanode, and then inside you will see a WrappedNode Input and WrappedNode Output, along with your other wrapped nodes:
Double-clicking on either of those gives you a configuration dialog where you can configure what flow variables get passed in and out of the wrapped metanode.
Thank you Steve! I’ve tried this a few different ways now with my Uniprot DB and am still trying to concoct the secret sauce that results in a useful table. Do you have a recommendation on Chunk Size for this? It defaults to 50 and the description mentions “larger values” but gives no suggestion of what a large value would be for this. I’ve tried 50, 100, 500, and 20 but the best I’ve seen so far is that they can cause erratic display behavior on my Windows system (while not completing parsing of the file).
OK, another thought - if you put the FASTA Sequence extractor node within a ‘chunk loop’, with a chunk size of say 1000 rows per chunk, does it ever complete?
And follow on question - are you able to share the FASTA file causing the problems, or is it proprietary? (or can you give an example of how to create something of similar magnitude to test with?)
To answer the first question, I see that the chunk loop causes it to get to 99% much more quickly. However it still seems to just stop there and do nothing. That said, I don’t usually use chunk loops in this way so I can’t be certain that I didn’t do something wrong here. I tried both ‘Chunk Loop Start’ and ‘Parallel Chunk Start’, and both very quickly hit 99% on FASTA Sequence Extractor and then promptly started doing nothing at all, with no sign of progress towards the end node of the loop.
As for your second question, this file came directly from Uniprot. I reached out to the PI who gave it to me, he confirmed that there is nothing proprietary in it and I can certainly share it with you if you want it. It is the Uniprot SwissProt Human Canonical Isoform set from May 27 2018. I’m not sure that I can upload that large of a file (~28MB) to the forum directly but I could find another way to get it to you if you’d like.
I just downloaded the Uniprot Human proteome again to make sure there wasn’t something specific to the file I’m using, and found that this file has the same behavior. The original file is ~33mb, I zipped it to 17mb which is still too large to upload here. The following link will lead to the file
Just a thought - did you select the ‘Delete FASTA Sequence column’ option? If not, then all 42k output rows will contain a copy of the whole 468k lines FASTA file, which will 1 - fill up your hard drive pretty effectively, and 2 - take a crazy amount of time to ever write the table to disk!
There maybe other issues with the way the node is written which come into play when handling files of this sort of size - I will take another look at that too, and it maybe possible to write a FASTA reader which solves all of the above problems at once without very much effort at all, similar to the other ‘Load … files’ nodes we have (e.g. this one - https://nodepit.com/node/com.vernalis.knime.io.nodes.load.xml.LoadLocalXmlFilesNodeFactory)
That option might be the solution I needed! It was never clear to me what that check box was going to do, so I always left it unchecked. I just went back, checked it, and it completed!
I was expecting that checking that box would give me output without sequence (which was the biggest problem I was having with other tools in that they were producing tables without protein sequences) so I never tried it.
Thank you!
Lee
I must admit I looked at that option and hoped it did the right thing - it’s definitely a bit badly phrased! This node is one of the first few nodes I wrote, and was only ever really tried out with FASTA cells resulting from the PDB Downloader node output, which generally have only a handful of sequences at most. I will see if we can improve a bit on the node performance and the naming of options (“Remove input column” would seem more obvious with hindsight!), maybe document that large input files are going to need that option checked if they are ever to complete, and look into a dedicated FASTA loader node.
(And I guess my original answer about KNIME internals writing tables to disc was partially correct at least!)
Watch this thread, and I will post back if I make some progress with any of those things, and thanks for your feedback and patience!
@izaychik63 - thanks. We already have abstract node classes which form the basis of a number of readers for text-based files (XML, SDF, CDXML, PDB, plain text etc), so we simply would need to create a couple of new implementing subclasses to add FASTA to the set in this case.
The Load FASTA files node is now available in version 1.18.0 of our community contribution (nightly build only at the moment - stable builds should hopefully follow next week)