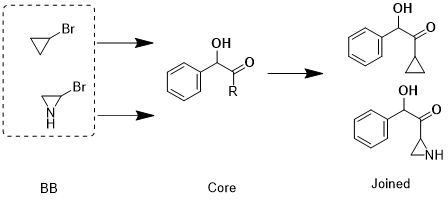
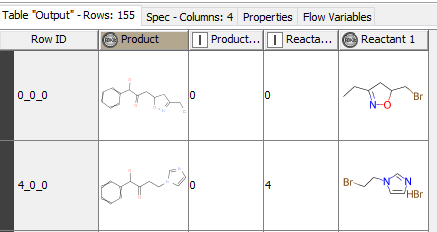
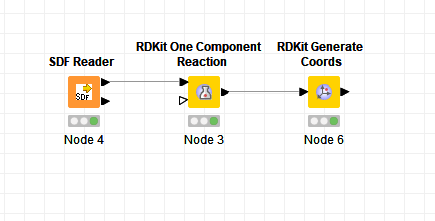
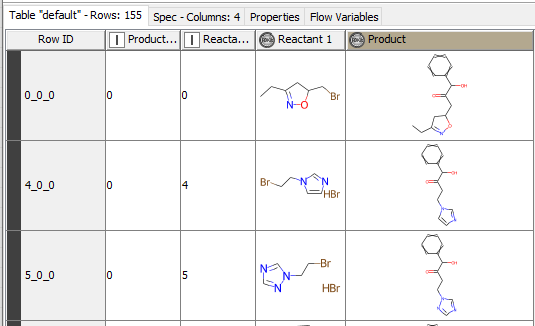
I am trying to filter the building blocks(bb) which contains only alkyl bromines not aromatic. Next use these number of bb to attach to the specific R-group of the core compound.
I have used SDF Reader to read both bb and core molecules >RDkit From Molecules-> RDKit substructure filter used smiles of br followed by RDKit Functional group. I would appreciate if you can suggest the steps for bb filtration and attaching to R-group.
Actually I did followed the Example server and was able to filter the building blocks with RDKit Functional Group Filter. However I am not able to attach these filtered fragments to the core molecule.
First I would like to get aliphatic bromines, next attach these fragments(Br as attaching point, by removing br) to the R-group of my core structure.
It should be something like this;
BrC1CC1 (building blocks) attach to OC(C([R])=O)C1=CC=CC=C1 and get OC(C(CC1CC1)=O)C2=CC=CC=C2
What I am really looking here as follows; Br containing bb attach to R-group in the core structure. Your suggestions could work with rxn within the bb, but I need rxn between R-group of core and bb. Hope my questions is clear here! Also, I wonder how do we make the SMART rxn scheme for this joining? Thanks
Your suggestions could work with rxn within the bb, but I need rxn between R-group of core and bb.
I’m afraid I don’t see the distinction you’re trying to make.
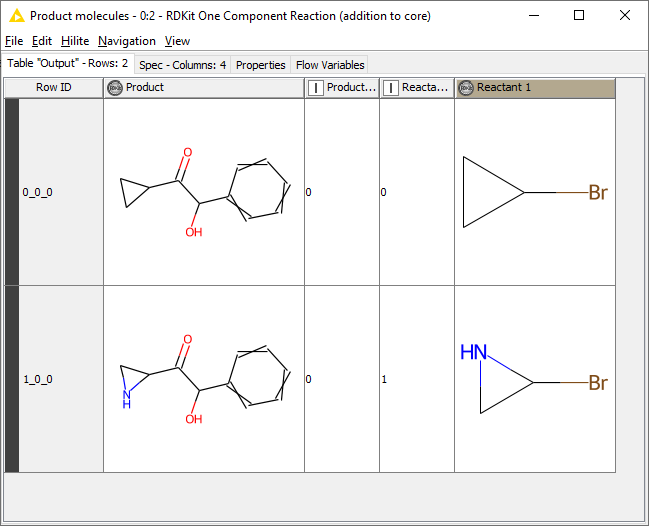
The workflow I shared takes the building blocks and substitutes the Br with the core. If you’re reacting each building block with the same core, then this approach works fine and gives the desired results. What exactly is the issue?
Also, I wonder how do we make the SMART rxn scheme for this joining?
I don’t think I understand what you’re asking here. I shared the link to the SMARTS documentation in my last post. It shows what these strings look like. If you’re familiar with SMILES then SMARTS is similar.
Can you show me how RDKit One-Component Reaction node works for this? Attaching two fragments is more like two component reaction? rxn = AllChem.ReactionFromSmarts("[Br][:1].[R][:2]>>[:1][:2]")?
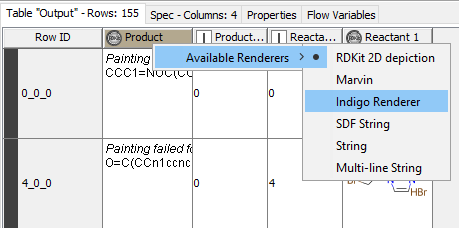
I tried something like below in RDKit One-Component Reaction SMARTS but didn’t works for me as it shown painting failed for error in product"
[#6:1][Br:2] >> [#6]-[#6]-#6:7-#6:6-[#6:9]-1=[#6:10]-[#6:11]=[#6:12]-[#6:13]=[#6:14]-1
Can you show me how RDKit One-Component Reaction node works for this?
I don’t quite understand what you’re asking for. The workflow I uploaded should have an RDKit One Component reaction node that carries out this reaction. Does it work when you download and execute the workflow? Have you looked at the node configuration?
Attaching two fragments is more like two component reaction?
From my point of view (and based solely on what you’ve posted so far) this is a simple one-component reaction: for every building block, you’re substituting the Br with the same chemical entity which you call the “core”.
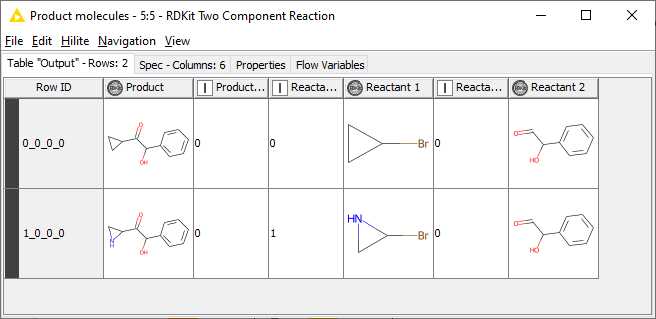
If you’d like a more complicated approach, you could use the two component reaction node.
I’ve updated the workflow that I previously uploaded to include this. Download it and look at the molecule tables and the reaction node configurations to see how they work.
I tried something like below in RDKit One-Component Reaction SMARTS but didn’t works for me as it shown painting failed for error in product"
[#6:1][Br:2] >> [#6]-[#6]-#6:7-#6:6-[#6:9]-1=[#6:10]-[#6:11]=[#6:12]-[#6:13]=[#6:14]-1
Do you have any experience with SMILES or SMARTS? Just by looking at it you should be able to tell that this product is not a valid SMARTS string. Where did you get it from?
There really aren’t any detailed steps that I can share. I created SMARTS for the reactants and the products (using a combination of intuition, ChemDraw, and the SMARTS tool I linked to earlier), then tried to assemble a reaction query that made sense. This type of work requires a lot of reading along with trial and error.
The SMARTS were copied from Marvin. I did used SMART plus and was having same issue.
The example workflow you provided works fine by reading the molecules from Table creator. However if I read bb from SD Reader->RDKit From Molecules → RDKit one component reaction gets “painting failed for”.
Do you want a column that contains the name of the building block that went into the reaction for that row? If so, then you’ll need to join the output of RDKit reaction node with the output from the initial SD file. If you generate a counter on the SD file table, you can do an inner join on the Counter column and the Reactant Sequence Index, then retain whichever “name” column you want from the SD table
Do you want IUPAC names of the final compounds? Unless you purchase a license for the ChemAxon nodes, there’s no way to generate IUPAC names from chemical structures.