It seems the Feature Selection Loop Start node using a GA selection strategy goes beyond upper bound number of features when BOTH lower and upper bound numbers are set to the same number. I am trying to keep the gene size the same in all cases.
Background: I want to use a GA to feature select in a NN train/test workflow. As we discussed before Keras Network Learner nodes struggle with any changes in the number of inputs, so to avoid this (to start!) - I simply want to keep the same number of input nodes, but use a GA-feature selection loop to identify a good set of input features for a particular input-output mapping.
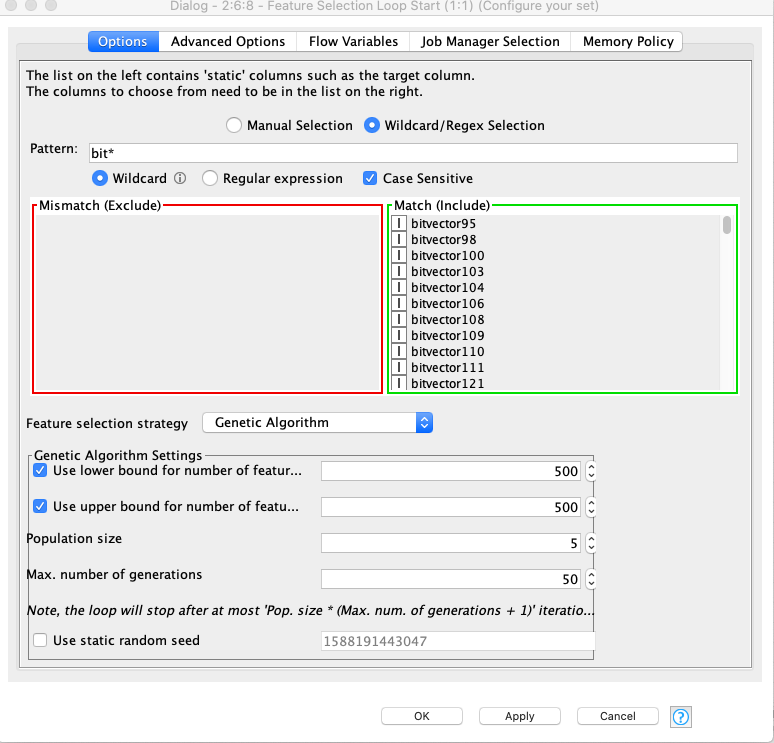
The feature selection dialog is below. My understanding is that this would keep all the gene’s of length 500. While this runs for seemingly the first 5 members (i.e. population), the next generation seems to have cases outside of the 500 range. This of course stops the workflow given the Keras component that follows in the workflow. The log entry indicates the GA actually provided 502 (not 500!)
2020-04-30 16:54:01,572 : WARN : KNIME-Worker-62-Partitioning 2:6:6 : : Node : Keras Network Learner : 2:6:495 : Selected input columns provide more elements (502) than neurons available (500) for network input ‘input_1:0’. Try removing some columns from the selection.
Is this a bug or a “feature” Any suggestions with a work-around, or if my understanding of the node is off, please clarify.
I did some investigation. This is, indeed, some kind of feature instead of bug:
The creation of each individual is done randomly and then validated. The validation checks, if lower and upper bounds for number of features are correct. If the validation returns false, a new individual is generated. This iteration limited to be done at max 100 times to prevent that the algorithm gets stuck. After 100 times returning false, any individual will be accepted.
This happens in your case. I guess we should make this parameter configurable (internal reference: AP-14133). 100 seems to be not enough for 500 features.
As a workaround, you can filter the columns manually by the best score achieved with exactly 500 features. See this workflow as an example: filter_features.knwf (25.8 KB) Note that you might need to adapt the settings of the Top K Selector node if you are using an objective that is minimized.
Thank you for looking into this. I definitely would call that unexpected behavior! At least as designed, the node continues and doesn’t halt on this failing. Good suggestion regarding filtering total number of features to meet your desired range.
Yes, makes sense to later add some clarity re: number of checks before fail and continue if fail, or stop.
Incidentally, since it was an immovable wall, I wound up leveraging the previous suggested approach to reforming inputs into the Keras network modeling to enable a more robust workflow. This has given me further perspective on the GA/feature selection node.
Actually, the node does NOT seem to well interrogate within the lower and upper bounds (in terms of features to select).
Any suggestion how to broaden this? For example, for a range of 100 (lower) to 1024 (upper), it seems to largely hover in the 500’s the whole time, never leaving the midsection much.
Did you play around with the parameters? Having a bigger population and different crossover/mutation settings might improve the exploration. Are just the number of features similar for different individuals or also the selected features?
Thanks Simon - I did experiment with different settings - e.g. single, double, uniform, also others - e.g. tournament v. Roulette, etc.
I had assumed mutation rate would not change length of genes (I.e. number of features). Was I incorrect? I could always try increasing that.
I haven’t studied closely which specific features were selected, but only noticed no substantial change in gene length - which I would expect with a big range in min/max. I am trying no min and no max now, but so far lengths are still about 500ish. Very odd.
The length of genes always stays the same. It is the number of available features. What matters is how many of the bits of a gene are 1s and how many 0s. The number of 1s define the number of selected features, so a mutation can change this. However, I wouldn’t set the mutation rate too high.
Would still be interesting to know if the 500ish features are always the same ones. Might be that about 500 specific features in your feature set are much “better” than the rest and the algorithm converges, although of the exploration, always to this point.
There are about 11K total features (some statistics on selection counts below), though I have also had just 1K with the same kind of behavior. I have a list of features and prediction correlations over several runs now, so I can analyze to answer one of your questions. Would be great to pull out the most frequently selected features from the list for the highest prediction correlations and experiment from there. Do you know of a node or workflow that can facilitate the analyses of a string of features?
The output from the feature selection loop effectively produces a table like this (bitcount, prediction corr, list of selected features":
Thanks @smuskal for the investigation.
During initialization, the probabilities of a feature being selected is set in a way that roughly min + ((max - min) / 2) features are selected. That means, having min as 100 and max as 1024 should lead to an initialization with about 550 features selected.
When you don’t set any limits and use 11k total features, the mean is about 5500. This is reflected by your statistics. So it seems that the initialization is working so far.
How big the shift of number of features during the execution of the algorithm is, depends on the features combined with the objective that is optimized and on the algorithm parameters.
Here is an example how you could count the number of occurrences of each feature in the different feature sets: filter_features.knwf (59.4 KB)
Very helpful feature counting workflow, thanks so much Simon. I love self-contained workflows that work right out of the box so I can learn from them! The seemingly restricted change in total number of features is still something that is curious and troubling. I can’t seem to get them to grow out of the mid-point range very substantially. Perhaps the score/prediction values need to be more dramatically different?
That’s a good point but depends heavily on the feature space. The stochastic nature of mutation and crossover prevents that the number of features of an individual (usually) changes heavily from one generation to the next one. And even if it does but the score is not improving significantly, the stochastic nature of the selection will prevent that this individual will be kept with high probability over the next generations. However, if there is a gradient in the feature space as, e.g., each additional feature added to a feature set with size 500 improves the score, I would expect the algorithm to move into this direction and result in feature sets that are near to 700 (how near depends on the number of generations). If the scores are similar and no clear direction of improvement (fewer or more features) exists, the algorithm will explore less and stay near to 500 features. That said, I would say the behavior might be just as expected in your use case.
As a side mark: Rank selection is often used if the fitness, in your case the score, is very similar for different individuals. This will increase the probabilities for ones with higher scores to be selected. That might be useful in your case.
Thank you again. I am now investigating exponential ranking in the GA selection strategy, and then linear ranking. Still running the first population, so will probably not know if the total feature count varies for a couple iterations. I am not optimistic that the gene sizes will be changing that dramatically though. I think there is a fundamental constraint somewhere that is keeping the gene lengths roughly similar.
 Any suggestions with a work-around, or if my understanding of the node is off, please clarify.
Any suggestions with a work-around, or if my understanding of the node is off, please clarify.