Since a while I’m using the Molecule Widget (Labs) node to input molecules on WebPortal’s workflows. In general this node works great and it’s very valuable for KNIME cheminformaticians community. Nevertheless I discovered that it has problems in specific cases. When I set the format to SMARTS and I draw a molecules structure containing R groups the following error is triggered:
Error: Convert error! SMILES saver: Unknown atom attribute 6
I’m using KNIME AP 5.1 and Molecule Widget (Labs) node corresponds to Ketcher v2.7.2.
Does anyone know why this happens and if it can be fixed? Workarounds are also welcome.
Thanks for reaching out!
I can reproduce the behavior, but I’m not sure what’s happening under the hood. To us it looks like something related to ketcher itself, rather than its integration/implementation into KNIME. I thought SMARTS don’t have R groups, so could it be that it’s an unprecise/unspecific error message?
To investigate it further it’s maybe also worth asking ketcher directly: epam/ketcher · Discussions · GitHub
Sorry I can’t help much there, but maybe somebody else in this forum knows more
Hi @Alice_Krebs,
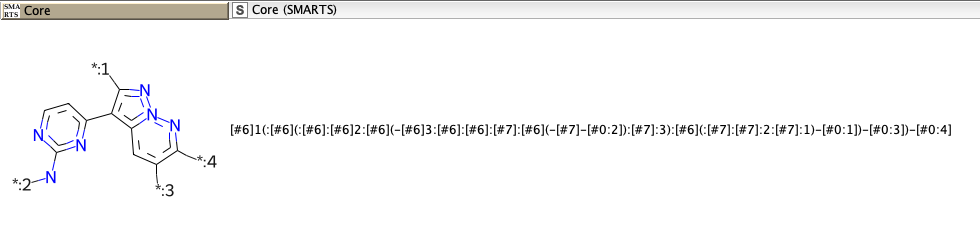
Thanks for your quick reply! I may be wrong about SMARTS containing R groups. What I would like to get is a SMARTS that represents a molecular structure core with substituents in different positions. For example, using RDKit to perform R-group decomposition will result in a molecular structure core expressed as a SMARTS where the substituent attachment points are defined as [#0:1], [#0:2], [#0:3], etc. (see figure below).
Ideally I would like to get a similar SMARTS using Ketcher and I thought the correct way to do this would be by using R-group numbering, but I may be wrong.
Do you have any alternative suggestions for me to achieve this?
Thanks for any advice.
I normally recommend that people use mol blocks (ideally V3000 mol blocks) as the output format from sketchers. This is particularly true for queries (your scaffold is intended to be used as a query).
V3000 mol, and the way the RDKit interprets it, almost always does a better job of behaving it the way you expect it to when used for searches.
I think if you give that a try, you’ll find that it interacts better with the r-group decomposition node.
If you really want SMARTS for some reason, it’s usually a better idea to read the V3000 mol into the RDKit and then ask the RDKit to give you the SMARTS for that molecule than it is to ask the sketch to give you SMARTS.
Hi Greg,
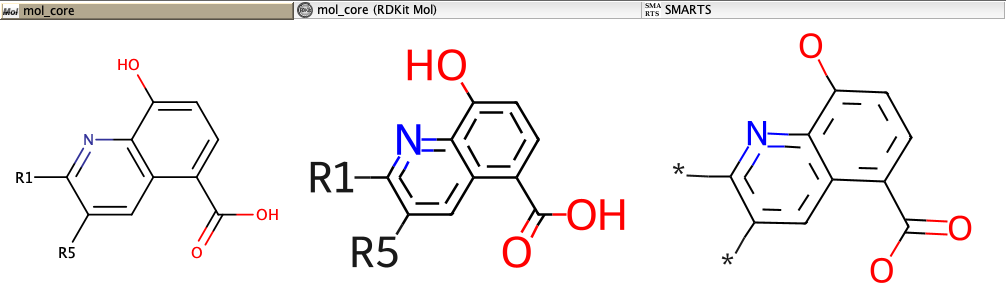
Thanks for your help here. I tried outputting a core molecule as a V3000 mol and then using RDKit to translate this into a SMARTS as you suggested, but it didn’t work. The numbering of the attachment points is removed in the translation (see screenshot below).
I will try to explain better what I am trying to achieve. As you can see in this workflow from Daria, the 2 inputs for the molecule enumeration step are:
A core molecule expressed as a SMARTS (e.g. [#6]1(:[#6](:[#6]:[#6]2:[#6](-[#6]3:[#6]:[#6]:[#7]:[#6](-[#7]-[#0:2]):[#7]:3):[#6](:[#7]:[#7]:2:[#7]:1)-[#0:1])-[#0:3])-[#0:4]) with numbered attachment points (e.g. [#0:1], [#0:2], [#0:3], [#0:4], etc)
A table of sidechains expressed as SMILES with numbered attachment points (e.g [H][*:1].COc1cc(C(F)(F)F)cc([*:2])c1.COCCO[*:3].[H][*:4], [H][*:1].Clc1ccc([*:2])cc1Cl.COCCO[*:3].CCO[*:4], etc.)
Here I’m wondering how I can obtain the first input from a molecule sketcher so that the molecule core and the attachment points can be defined by a user. Is there a way to do this with KNIME?
Ah, got it. That workflow from Daria (by the way: I really like that one. We had a lot of fun putting together.) is looking for atom maps on the dummy atoms (or R groups) in order to know where to attach things and you don’t have atom map information on the scaffolds you’ve drawn.
I know that ketcher can do atom mapping for reactions, but I failed to figure out how to make it work on molecules. This means that a bit of a “work-around” (a.k.a. “hack”) involving a string manipulation node is required.
The attached workflow shows how to modify the V3000 mol text that comes out of ketcher and add atom maps to each of the labelled R groups. The RDKit can read this information and it is translated into the output SMARTS.
Hi Greg,
I’m sorry if I was not clear in my previous message. Your hack works perfectly. I’m not familiar with the V3000 mol format so I didn’t know what to work on. Maybe is time for me to learn about it
Thank you very much for your help and viva RDKit!
Hi Evert,
Thanks for the tip. Unfortunately I don’t have a license for MarvinSketch. Instead, the solution proposed by @greglandrum is completely open source (Ketcher + RDKit), so it can be used by anyone.
Best,
Gio
Evert,
Thanks for the info! I didn’t know that Marvin could work without a licence.
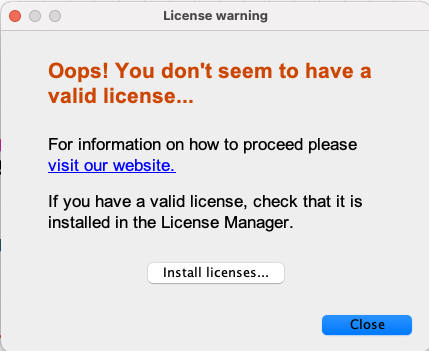
I’ve the license for some JChem extensions but not for Marvin. I had previously installed Marvin from the KNIME Partners Extensions repo. I’ve uninstalled it and reinstalled from the URL you suggested, but I still get the following message:
That’s weird, isn’t it?!
Best,
Gio