thanks all for your interest and good feedback,
I will try to answer in one post:
@linh
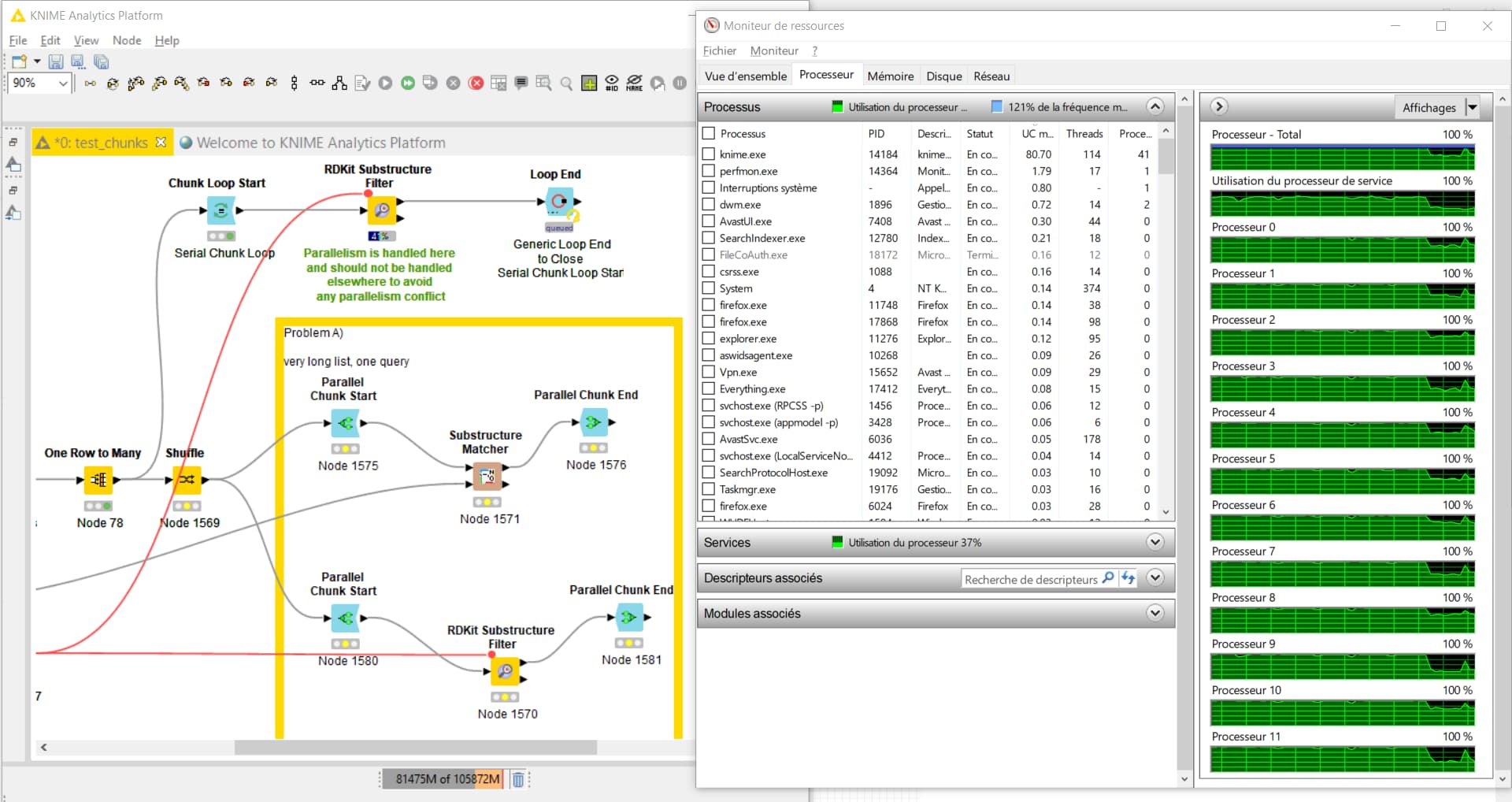
I use the KNIME GUI in an X-Session of a 192-cpu server.
I alreade checked the threads and increased the memory in the knime.ini by changing
-Xmx2048m
to
-Xmx16g
can I do any further tweaks using the knime.ini?
I don’t know what a jstack is, but I shared an Example workflow further below.
@kienerj
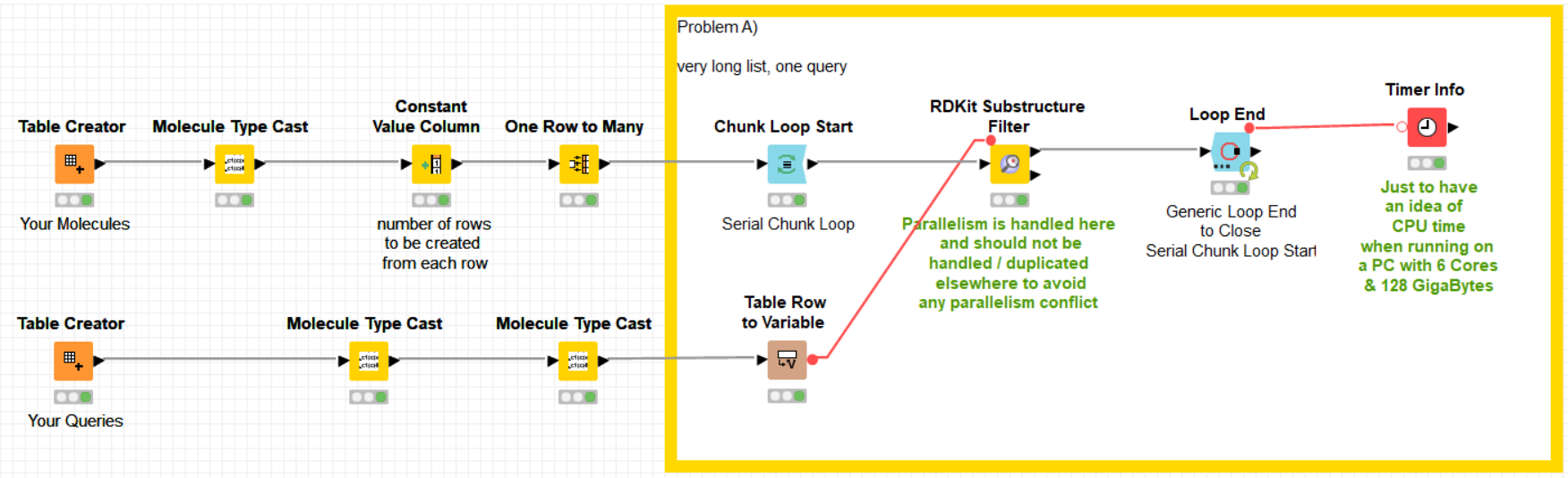
my two main application is filtering of chemical molecules using the “substructure matcher” node. In this case 4.5 billion of rows with molecules are compared to 1 query molecule. So the operation on every row takes only a fraction of a second, and the rows are independend, thus the table can be split by parallel chunking etc. No matter if I split the table beforehand to 50 parts or use a parallel chunk, KNIME will never use more than aprox 20 CPUs at a time. This operation takes days then  … from the “computational effort” this should be doable in a few hours or less with 192 cores.
… from the “computational effort” this should be doable in a few hours or less with 192 cores.
So if I take one node, KNIME uses 2-3 CPUs and gets 300k rows done in 1min. As I have 60x more CPUs it should be doable in a second per 300k (on average), thus the whole shabang should be over in 2-3 hours.
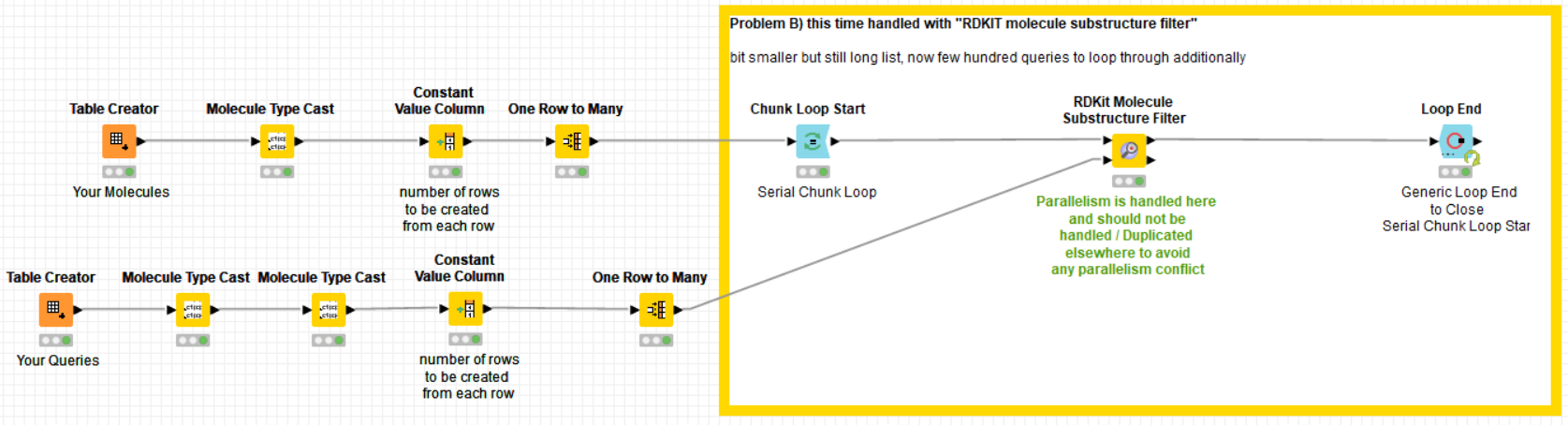
Funny that you mention RDKit, my 2nd application is with their “Molecule Substructure Filter”, which, same as the “substructure matcher” mentioned above, is not multi-threaded, unfortunately. Takes about the same time as the substructure matcher but is more difficult to use due to the format requirements of the input, queries need to be variables etc.
(I noticed when the chunks are set very high, e.g. 250-300, then I see in case of the RDKit matcher some spikes where 30-35% of CPUs are used … but still by far not the “full force” )
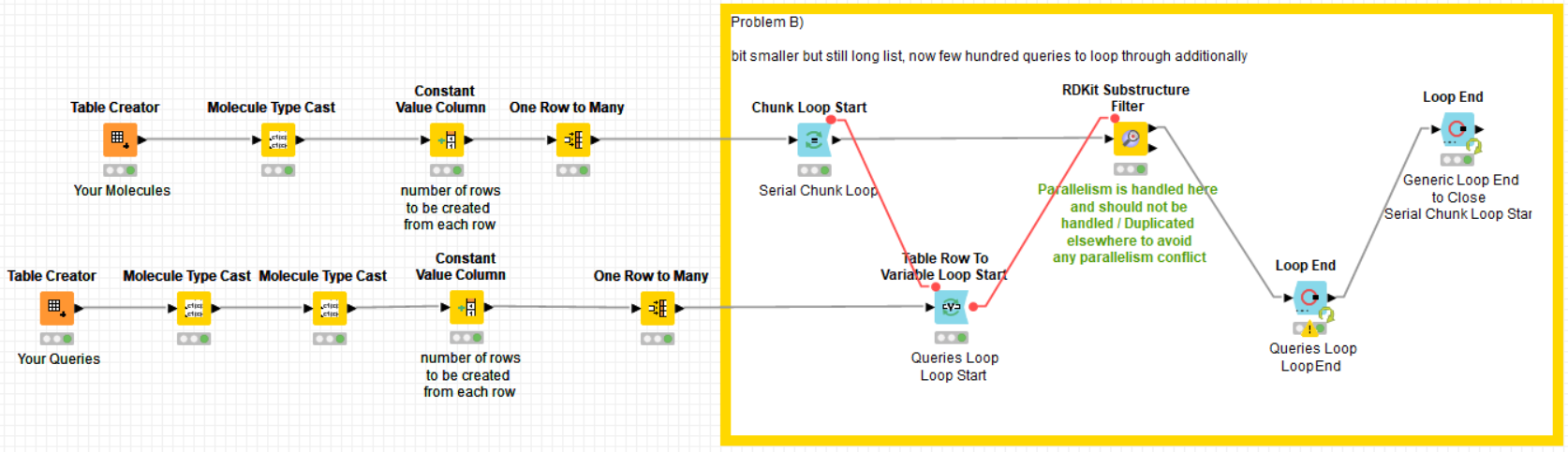
Anyway, in my 2nd case I have a list of 1-10 million molecules (rows) and match them to 200-700 queries.
Also here splitting the table or using parallel chunks does not get KNIME to do things in parallel and it takes a whole working day to run this calculation. Should be doable in 30min or so with 192 cpus.
@Daniel_Weikert
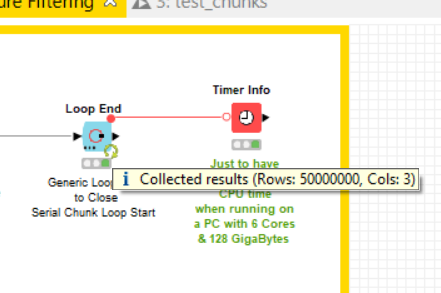
as I cannot upload the 130GB of data for the 4.5 billion rows (SMILES format), I simulated this with a “one to many rows” node. I did three typical examples in there, 2 with one query and one with 500 queries. maybe this helps.
here the example workflow:
test_chunks_.knwf (59.8 KB)