I have downloaded your workflow and made a few modifications to make it work.
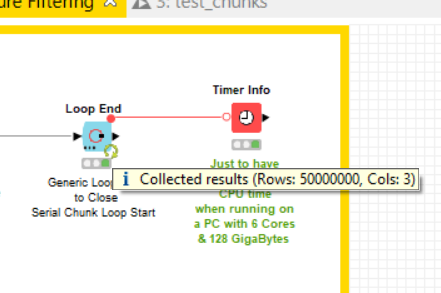
I have run the workflow on a laptop with 6 cores (12 threads) & 128 Gigabytes memory and it took 10 hours to run up to successfully execute your problem A) with the same number of rows (100 Million). The solution implemented is the following:
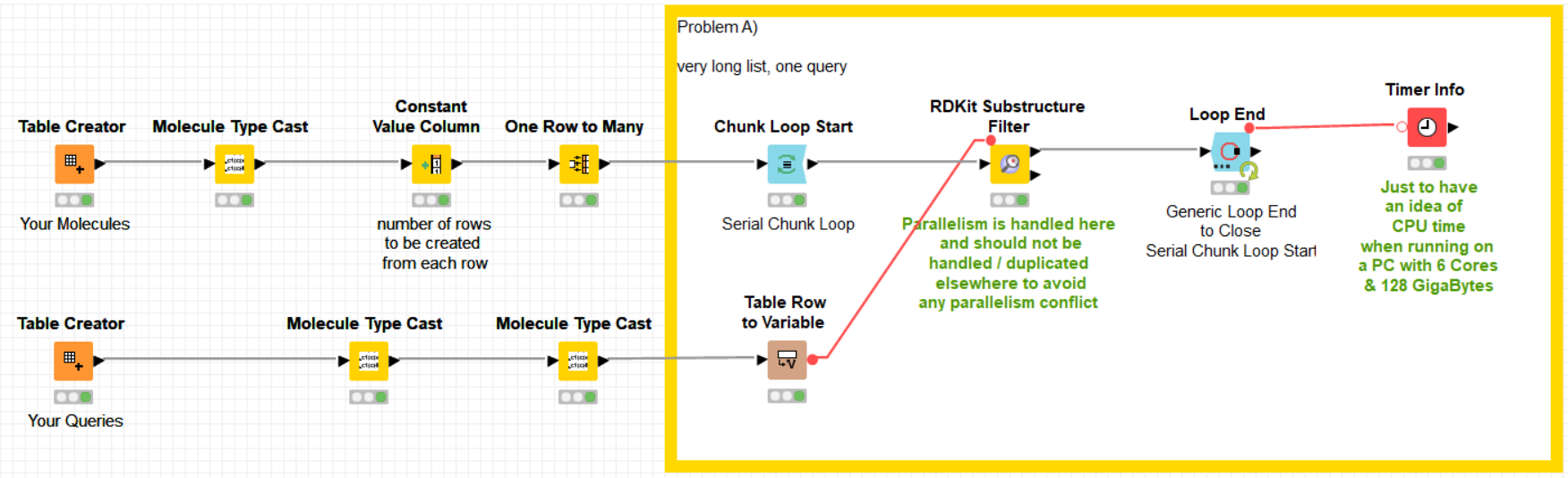
As one can see from the image, all the parallelism is achieved by the -RDKit Substructure Filter- node. This node handles itself the parallelism and does not need any further parallelism to be added around it. In fact, if parallelism is added using -Parallel Chunk Loop- nodes, then the two parallelism schemes fight each other against resources and this is most probably what was happening with your implementation. In other words, it is not recommended to encapsulate two parallel solutions because it generates competition for resources. It is neither recommended to run in parallel two parallelized branches in a workflow for the same reason.
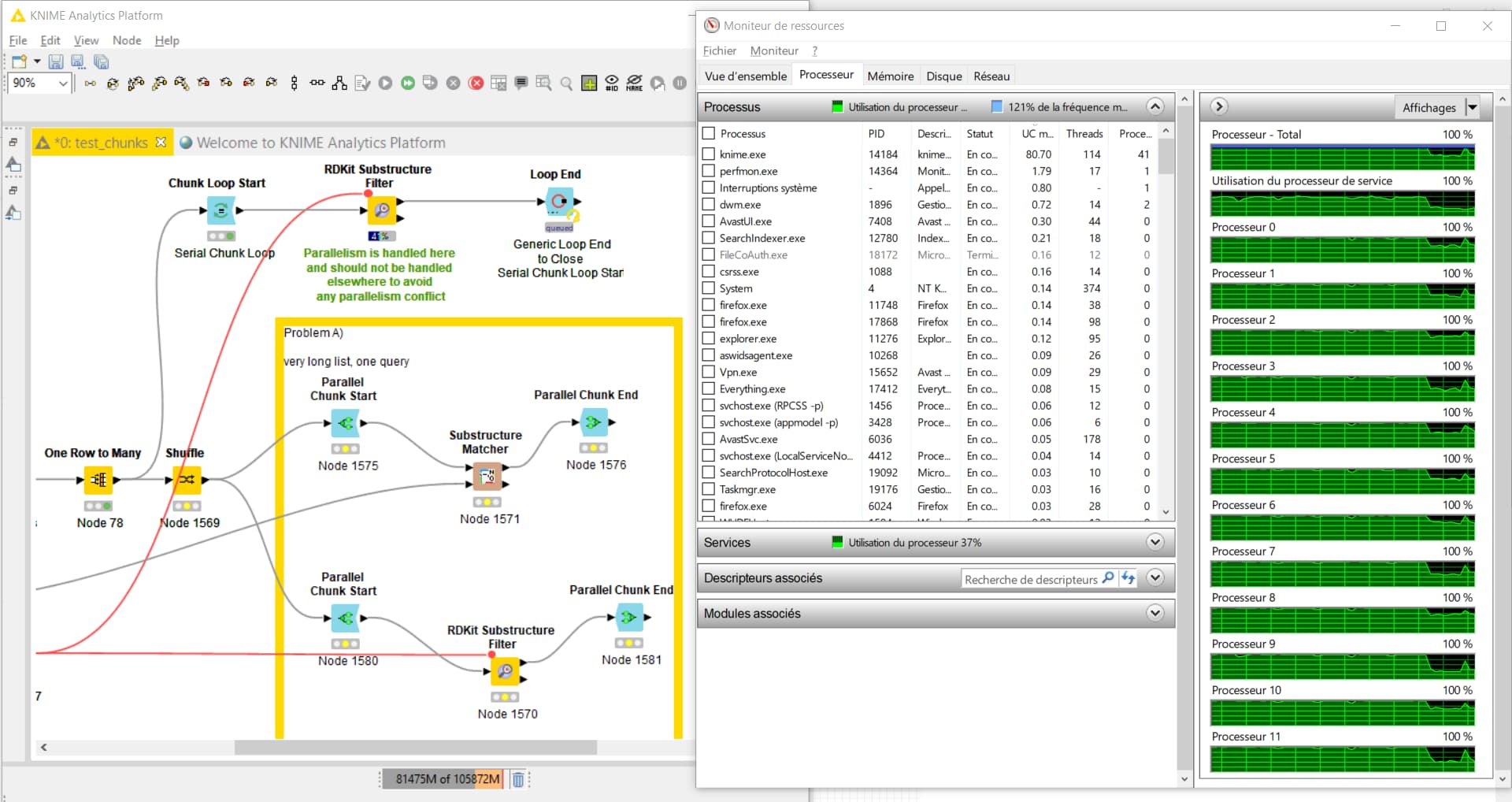
As you can see below, the solution is fully using all the cores in my computer.
I have modified “Problem B)” in the same way to show how to implement a possible solution too. It should work with your initial configuration too and much faster than in my PC given the hardware of your server:
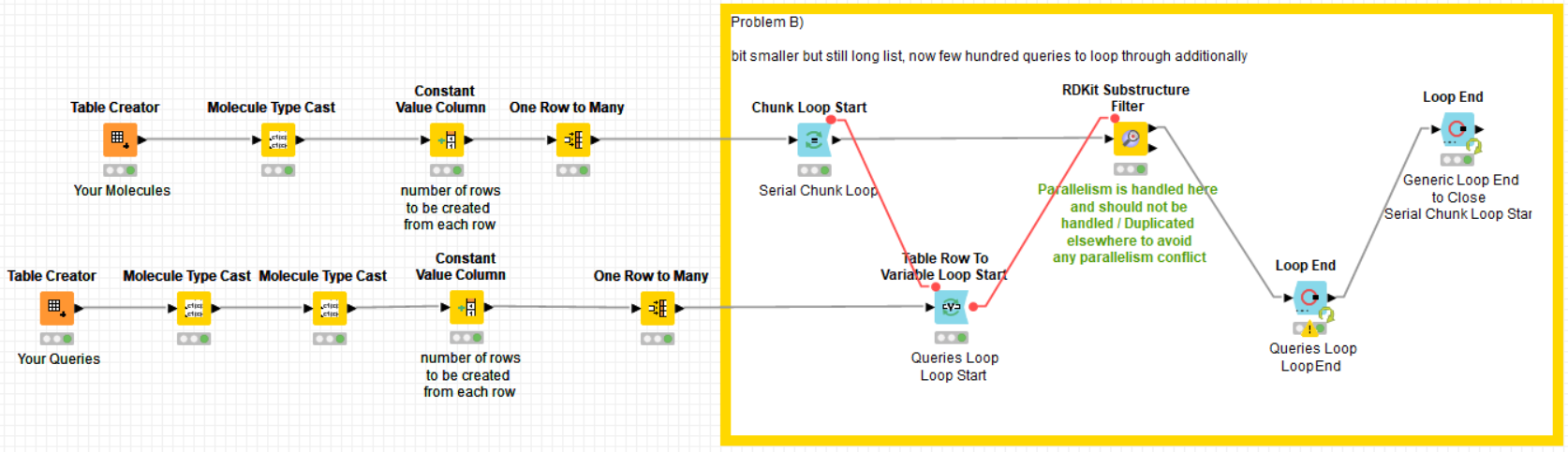
Still there is the question of why your server only runs a subset of its cores when you execute your workflow. Maybe it happens because it has been configured in this way by its administrator to limit the maximum number of processes per user. Nevertheless, the solution I’m providing here should work quite faster in your server and even better now that you have extended the memory upper limit.
The executed workflow can be downloaded from the HUB here:
The uploaded workflow has been configured and executed only with 10 Million molecules to limit the total amount of occupied disk in the hub but it has been verified and executed locally on 100 M.
Hope this helps. Please reach out again if you need further help.
Best
Ael