Hello everyone,
I’ve just realized that the RDKit Add Conformers node can change the chirality of stereocenters even when the “enforce preservation of input chirality” option is selected.
In the attached workflow 200 conformers are generated starting from a single molecule containing a defined chiral center (C1N(C)C[C@@H](OC2C=CC=CC=2)C1) and using the default node options, among which the one to “enforce the preservation of input chirality”. However, if the 3D structure of such conformers is visualized, it can be seen that the conformers in row1 and row39 have the chiral center inverted (i.e. “R”) respecting the one of all the other conformers and also the input molecule (i.e. “S”).
The chiral center inverstion can also be seen in the change of the second block of the generated inchikey, which is QTFVFOQCSACSKY-LLVKDONJSA-N instead of QTFVFOQCSACSKY-NSHDSACASA-N of the starting molecule.
Oddly enough, when the conformers are exported as SMILES, they are all exported with the same correct stereo center.
Can anyone confirm if this is a bug? Does anyone has any suggestions or comments?
@LunarJourneyer, thanks for your prompt reply.
Unfortunately I don’t think it’s just a visualization problem for at least 2 reasons:
As I explained in my first post the InChI keys generated for the conformers (using RDKit To InChI node) differ in their stereochemistry layer (please see the workflow attached to my first post).
In the exported sdf file, the atom parity of the chiral center is “1” in some molecules and “2” in others.
In my understanding this means that in some way the molecule is modified during the conformational search.
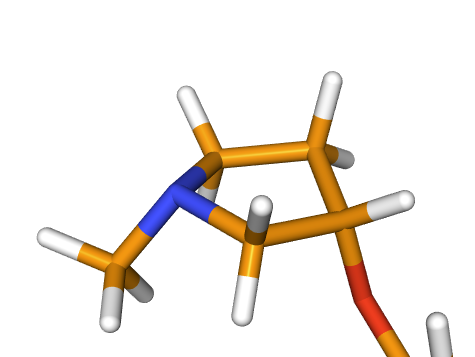
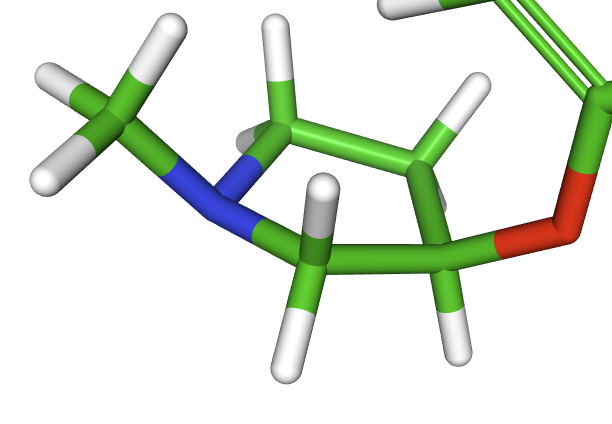
I attach here the 3D visualization of the ring containing the inverted chiral center. I tried to represent the ring from a similar perspective.
In the orange molecule (Row0) the configuration of the stereocenter is “S” as in the original one:
In contrast, in the green molecule (Row1) the configuration of the stereocenter is “R”:
Finally I think I have understood what the problem is and how to avoid it. As you can see from the workflow uploaded below, this problem only seems to happen if the molecules used as input have implicit hydrogen atoms (Hs). In fact, if explicit hydrogen atoms are added before the conformer generation step, the problem does not seem to occur. However, it is confusing that if this condition is a requirement, it is not explicitly stated in the RDKit Add Conformers node description.
To avoid potentially serious problems, it would be great if one of the next versions of the RDKit Add Conformers node would require the input molecules to have explicit Hs and refuse to run otherwise. This is exactly what happens in the RDKit conformer generation function when called from its Python interface.
Additionally (or alternatively) it would be good to mention that the input molecule should have explicit Hs directly in the description of the RDKit Add Conformers node.
I mention @manuelschwarze so that he is aware of this problem.