This workflow provides a simple example of generating matched molecular pairs (MMPs) from a set of compounds and using them to predict models with improved properties - in this case, CYP3A4 inhibition using ChEMBL data
Hi,
I’m trying to fragment the molecules of the example used in this workflow by selecting the “User defined” option of the “MMP Molecule Fragment (RDKit)” node.
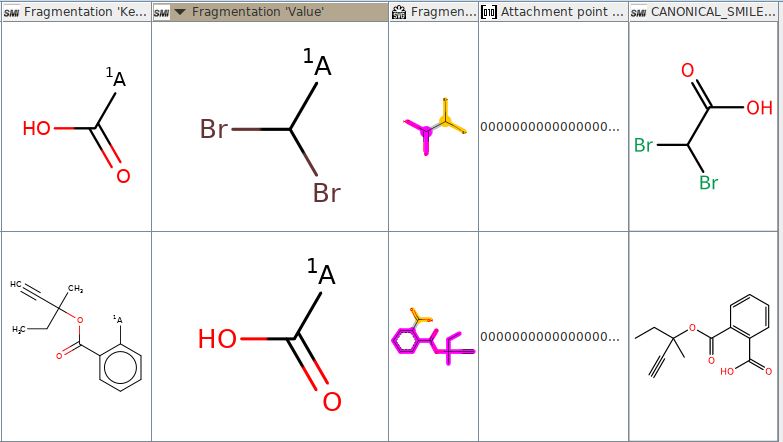
I would like to fragment the molecules in a way that as a result I obtain a carboxylic acid (-COOH) bound to whatever other atom. The rule I’m using it the following: [$([CX3](=O)[OX2H]):1]!@!=!#[*:2]>>[$([CX3](=O)[OX2H]):1]-[*].[*:2]-[$([CX3](=O)[OX2H])]
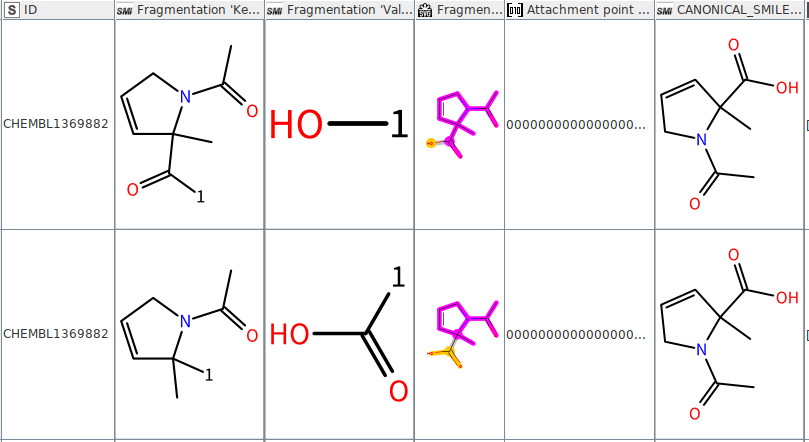
As a result I correctly obtain the splitting for the COOH (wanted) but also of the OH from the carboxylic acid (unwanted), as shown in the attached image. Is it a problem of the node or am I using the wrong rule? Thank you for your help!
Riccardo
@riccardomartini in this case, I think your rule is incorrect. The first atom matched by [$([CX3](=O)[OX2H]):1] will be the C atom of the COOH group, and the second is matching any singly-bonded match. I would be inclined to use the following pattern, which works:
[#6;$([CX3](=O)[OX2H]):1]!@!=!#[#6:2]
It will match aromatic and aliphatic COOH - you can adjust that with the second atom SMARTS if needed.
(NB The node no longer requires the full rSMARTS - just a two-atom matcher)
With your code I do not match the R-OH anymore. However, I’ve noticed that sometimes the carboxylic group is in the “Fragmentation ‘Key’” column instead in the “Fragmentation ‘Value’” column. Like in the image I attach here below. I can use a workaround of course, but I’m wondering if there is an more elegant solution that I’m currently overlooking.
I can’t think of a way to do it directly in the fragmentation node. A row filter should then be able to just keep rows with [1*]C(=O)O in the “Fragmentation ‘Value’”
(If you wanted it the other way round, then the easiest way to achieve this is via the Fragmentation Filtering Settings tab. Turn on the Filter by maximum number of changing heavy atoms option, and select a value of 3. Unfortunately, that’s not perfect either!)
Is there any tutorial or webinar or anything else which helps to work with or/and adopt the workflow? Could you please share some files or links to understand and to dive deeper into the topic?
There are no other materials available. The best approach would be to read the paper on the original algorithm which the nodes encapsulate:
J. Hussain and C Rea, “Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large datasets”, J. Chem. Inf. Model., 2010, 50, 339-348 (DOI:10.1021/ci900450m)
There is also a simplified description of the algorithm in:
S. D. Roughley, “Five Years of the KNIME Vernalis Cheminformatics Community Contribution”. Curr. Med. Chem. 2020, 27(38), 6495-6522 (DOI: 10.2174/0929867325666180904113616 (Open Access publication - no subscription required)
and then work through the example workflows to gain an understanding of how they are working, maybe adjusting some settings as you go. If you find specific settings that you don’t understand, please do ask.