I want to take this up in a new thread. Here my quote from the announcement thread:
I want to take up on this observation again which existed in legacy Python nodes and not is still in the new one.
Transferring rdkit molecules from KNIME to Python is very slow. See my quote above. It’s always a lot faster to use sdf or SMILES and do the conversion in the Python code.
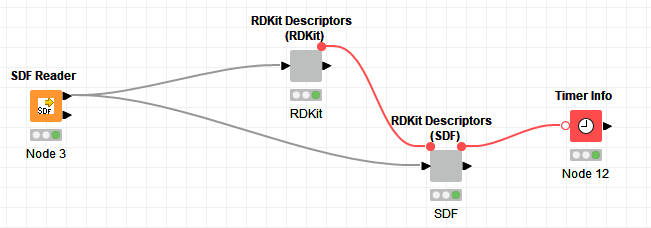
Case with RDKit uses RDKit from molecule before the python snippet, the case “SDF” just uses MolFromMolBlock in python code.
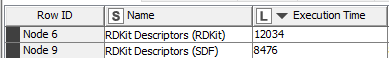
This is for about 3000 molecules. I put the python code at the end of the post.
I tend to go “out of my way” to input smiles or sdf into python nodes over rdkit mols for speed. It can really start to matter with higher number of molecules.
Anyway I was wondering why this great difference exists? I thought there is no more copying of memory with the new nodes but it somehow looks like some form of moving/copying data is happening?
Code with rdkit mols:
import knime.scripting.io as knio
from rdkit import Chem
from rdkit.Chem import Descriptors
from joblib import Parallel, delayed
import pandas as pd
def getMolDescriptors(mol):
'''
calculate the full list of descriptors for a molecule
'''
res = {}
for nm,fn in Descriptors._descList:
# some of the descriptor fucntions can throw errors if they fail, catch those here:
try:
val = fn(mol)
except:
val = None
res[nm] = val
return res
df = knio.input_tables[0].to_pandas()
descriptors = Parallel(n_jobs=6)(delayed(getMolDescriptors)(x) for x in df['RDKit Mol'])
desc_df = pd.DataFrame(descriptors, index=df.index)
output_table = df.join(desc_df)
knio.output_tables[0] = knio.Table.from_pandas(output_table)
Code with sdf/smiles:
import knime.scripting.io as knio
from rdkit import Chem
from rdkit.Chem import Descriptors
from joblib import Parallel, delayed
import pandas as pd
from rdkit import Chem
def getMolDescriptors(mol, mol_format):
'''
calculate the full list of descriptors for a molecule
'''
res = {}
for nm,fn in Descriptors._descList:
# some of the descriptor fucntions can throw errors if they fail, catch those here:
try:
if mol_format == "Smiles":
mol = Chem.MolFromSmiles(mol)
elif mol_format == "SDF":
mol = Chem.MolFromMolBlock(mol)
val = fn(mol)
except:
val = None
res[nm] = val
return res
mol_format = knio.flow_variables['Column Type']
df = knio.input_tables[0].to_pandas()
descriptors = Parallel(n_jobs=6)(delayed(getMolDescriptors)(x,mol_format) for x in df['Molecule'])
desc_df = pd.DataFrame(descriptors, index=df.index)
output_table = df.join(desc_df)
knio.output_tables[0] = knio.Table.from_pandas(output_table)