The previous workflow is ok, but, in the new workflow It is wrong.
WARN AccurateMassSearch 7:18 Invalid MIMEtype at port number 0 : has extension: ‘mzml’; expected one of: [featurexml, consensusxml]
Seems like a wrong connection. You need to connect to something that produces a featureXML, e.g. FeatureFinderMetabo
the metaboliteAdductDecharge in positive is this ,if in negative, should I change the potential_adducts.And as you said, if I use two Decharge node,one is positive,another is negative.
![]()
Usually per sample you only have one type of adducts, no? You are only measuring either in positive mode or in negative mode, aren’t you?
If you are measuring in mixed mode, I would probably split the file first.
For AccurateMassSearch, you can give both Positive and Negative Adducts (and it will choose the correct ones automatically), for MetaboliteAdductDecharger you need to specify the correct ones depending on the polarity.
Yes,I measure the two mode positive and negative pattern.
What I understand as you said is that I can deal with the positive mode although they are different group.
For MetaboliteAdductDecharger node,I need set the positive and negative adducts to group them,
So, there are positive and negative mode scans combined in one single file? Or in different files?
They are In different files,such as pos file,neg file. But, the file have different gioups.
With “groups” you mean potential adducts?
No,I am sorry I describe vaguely. It means different sample group.
Intringly,The AccurateMassSearch node appears error as before. I think I should limit the parameter. Oh ,but I forget the key changed.
Read 45372 entries from mapping file!
Read 6 entries from adduct file ‘D:\LIPIDMAPS\Adducts\PositiveAdducts.tsv’.
Read 7 entries from adduct file ‘D:\LIPIDMAPS\Adducts\NegativeAdducts.tsv’.
Error: Unable to read file (‘D’ found. Please use only valid element identifiers or modify share/OpenMS/CHEMISTRY/Elements.xml! in: Unknown element ‘D5’)
Can you post the contents of the Adducts files?
Is it?I change the adducts to analysis lipids.
M-H2O-H;1-
M-H;1-
M+CH2O2-H;1-
M+C2H4O2-H;1-
2M-H;1-
2M+CH2O2-H;1-
2M+C2H4O2-H;1-
M+H;1+
M;1+
M+NH4;1+
M+C3H8O;1+
2M+H;1+
2M+NH4;1+
This looks fine. Then the error is probably in your mapping files.
Yeah,I think it may be in the node.Because the error in the new workflow without MetaboliteAdductDecharger node is right. I will try it again.
Now I have a question that why I load the 8 data the output show 1 file.
I don’t how to distinguish them.
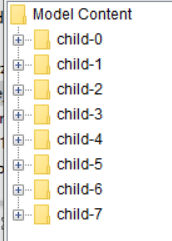
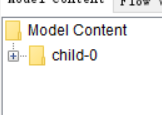
Does the workflow only excute one file?
The second picture is probably from inside the (Zip)Loop. There only one file is executed per iteration.
Yes,en,the one File appear in the node such as FileConverter.How should I check the Node
mzTabReader. Is it contain the all input 8 files? I also want to know how to look over the 8 files.
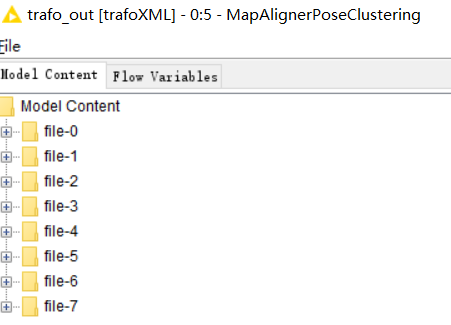

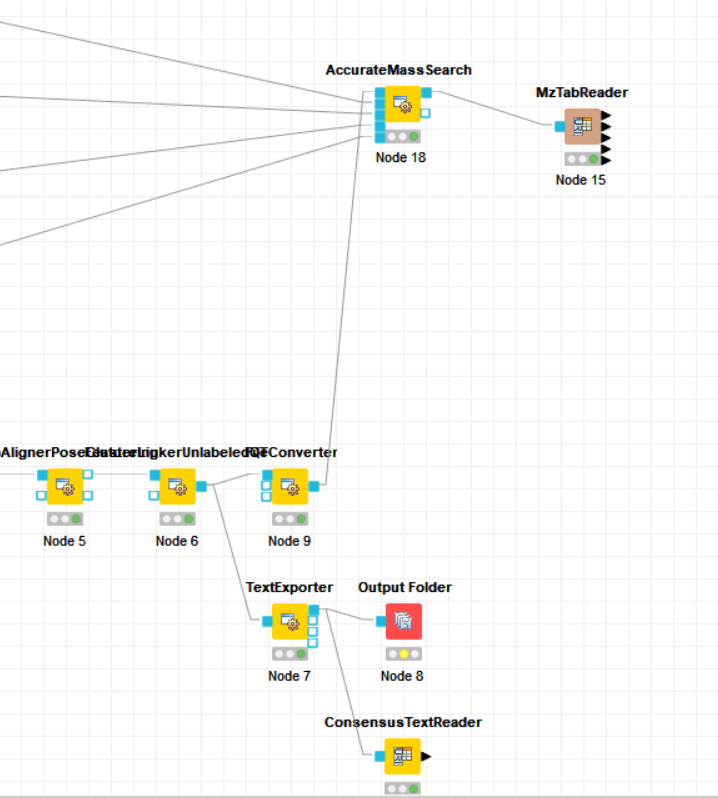
Yes, after FeatureLinkerUnlabeled, the data is combined into one file. MzTabReader later exports this data into a table format with several sections. You can view each section as a table by right-clicking on MzTabReader and selecting one of the output tables on the bottom of the context menu (in your case you will only have “Metadata” and the “Small molecule” section).
You can export the table data into a file by using e.g. a CSV/File Writer. Or you can export the original mzTab by placing an Output File node after AccurateMassSearch.
This topic was automatically closed 90 days after the last reply. New replies are no longer allowed.