Hi,
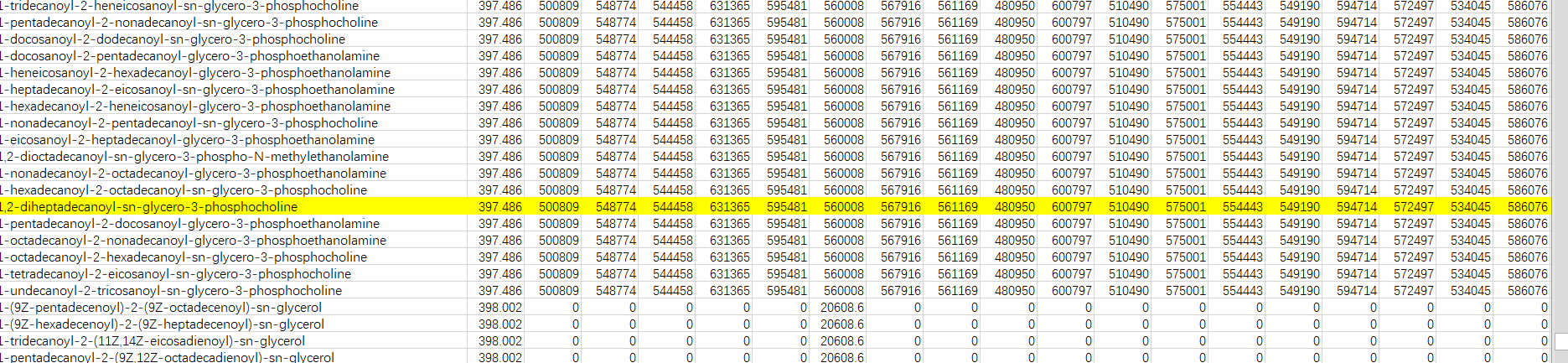
When I analysis the data, I need to consider to delete the repetitive row.
such as the internal standard marked yellow.Because I know it is it,I can delete others.However,other row like this repetition.What should I do?
Hello @zero,
Thank you for your question. In general you can automate the removal of duplicates in your data with your <duplicate row filter> node. In there, you can specify which column(s) should be tested for duplicates and what to do once they are found.
If you want to select your duplicates manually, you can use the <table view> node. It comes with an interactive view, in which you simply can select the desired rows. When you confirm your selection the node appends a new column with boolean values depicting which rows were selected (true) or not (false).
Does this help you with your analysis?
3 Likes
Yes,this is good for select the data,but I don’t know the standard of the removing.
Hello @zero,
In this case, can you please elaborate a bit more on what you are trying to achieve?
As shown, you either can automatically remove duplicates by using the <duplicate row filter> or manually remove it while using a combination of <table view> and <row filter> nodes.
Best regards,
Kevin
I want to remove the same retention time.Finally,it should be one compound corresponding to one retention time.
Hi @zero
Will the normal Row Filter node do the job there? Or the Row Splitter node, if you want to have the excluded data still available?
Although this is a good method, it seems not suitable for my result.
Thank you very much.
Hi!
How do you want to decide which compound to keep per retention time?
Hi,
I want to identify the compound with the other information.The method of the node I don’t know it is good for my result.
Hey,
This is unfortunately not possible. For further reduction of candidates you need specialized tools (preferably specific for lipidomics). I assume that even those will most often not give you ONE certain candidate.
You can maybe try it with spectral matching if you have any reference fragment spectra.
Ok,Thank you very much, I will use this workflow to identify the compoucd.But it excutes one sample at one time.Do you have some nice advice?
Thank you very much!
Hi,
you could use an Input Files node for multiple mzMLs and use a ZipLoop to loop over file ports.
If you want to append results from the MzTabReader in table form, you need to use InputFiles → PortToFileCells → ChunkLoopStart → FileCellsToPort → your workflow → LoopEnd.
Thank you very much, I will try it.
Hi,
I try it again use the node as you said,then it achieve it. The final is about this.From#0 to last sample.
I also find another use the workflow to identify lipid, but I get the pdf,not the workflow. It should use one file to analysis the data.

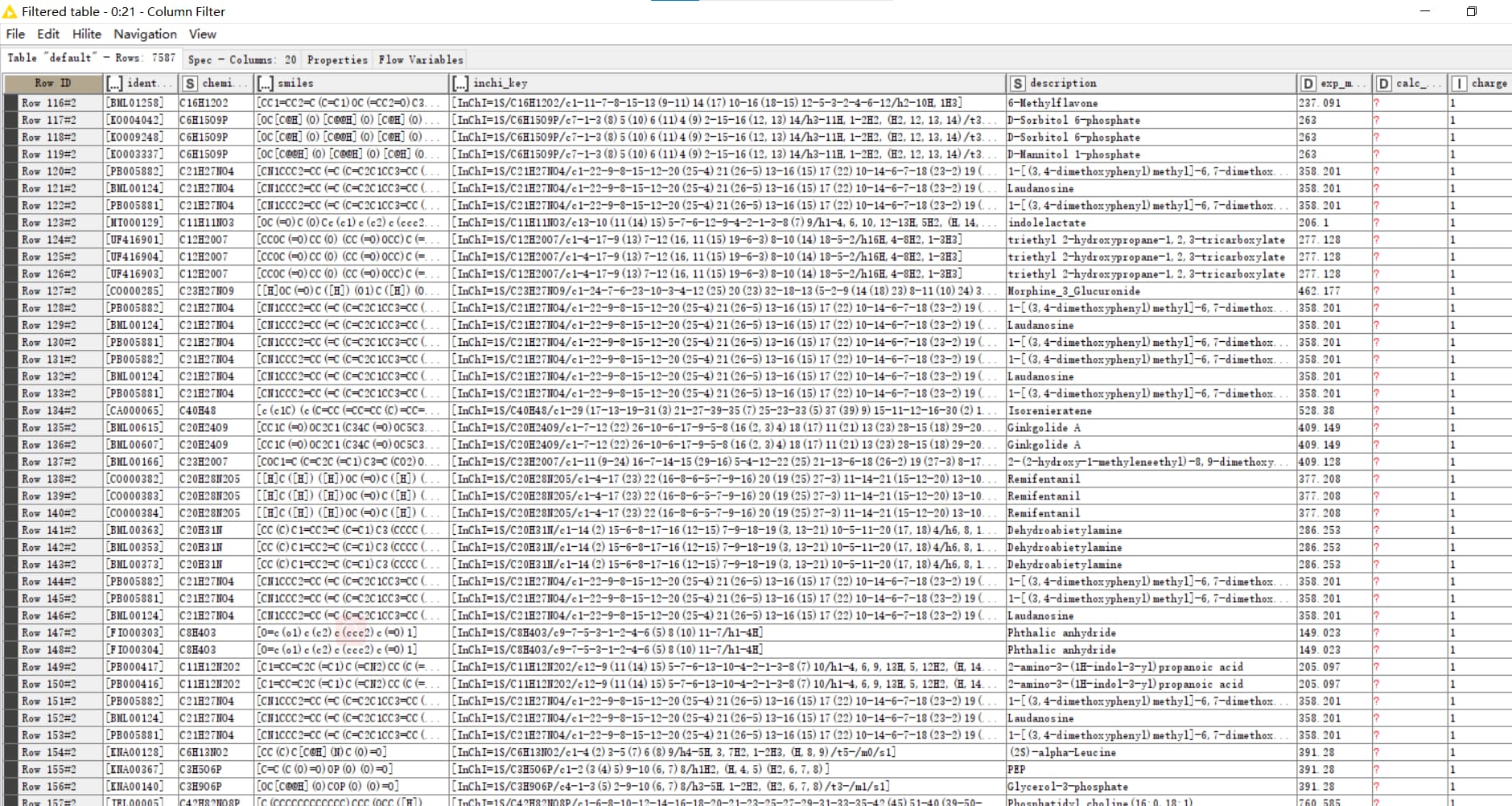
a) I want to see the joiner as above workflow. When I excute the joiner, it product an empty data.
b)Then if I should inspect the result AccurateMassSearch and the MetaboliteSpectraMatcher or not.
Thank you!
Hi!
Good start! I would reuse the mzML that goes into AccurateMassSearch and also connect it to MetaboSpectralMatcher.
Then for the Joiner: I actually think that this can NOT be solved with a simple Joiner. The SpectralMatcher will report identifications on MS2 level, while AccurateMassSearch will report identifications on MS1 level. You need to match with a certain tolerance. I don’t know of any KNIME node that can do that. You probably need a script.
Hi,
If your meaning is that the table from AccurateMassSearch node as input to connect the MetaboSpectralMatcher,I try it but it is not fluent.
Then,the script need I make some codes. I think it is diffculty for me as a freshman.
Thank you!
Hi,
I want to know if the two results from the SpectralMatcher node and the AccurateMassSearch node are the rt and mz of precursor ions information.But the information exists a little errors.
Yes, I think the problem is, that the SpectralMatcher has mz and RT of the precursor scan and AccurateMassSearch has the mz and RT of the “feature”/“3D peak”.
Therefore they will probably not match exactly.
This topic was automatically closed 90 days after the last reply. New replies are no longer allowed.