Thanks Iolanda - the NullPointerException was I think the problem which hopefully is now fixed in v1.27.0 upwards. Let me know how you get on with the updated version.
Steve
Thanks Iolanda - the NullPointerException was I think the problem which hopefully is now fixed in v1.27.0 upwards. Let me know how you get on with the updated version.
Steve
Hi Steve, unfortunately it keeps doing the same. I have now installed the version 27 and restarted KNIME. I kept 3D coordinates output only re-ran PMI node but it fails immediately, doesn’t show anything in the output table.
No sure exactly what I am doing wrong actually… the workflow is the same used for the small set with only the insertion of a col filter…
Are you able to attach a copy of the failed workflow and I will see if I can reproduce the error?
Thanks
Steve
Hi Steve, thanks! I now made a change in the configuration of the initial file which I found to be the only difference with the testset that worked. I am now calculating it again and calculating also another subset of 10000 non-confidential molecules that I can eventually share with you. I am waiting for it to finish (3D coordinates generation takes a while) and I will get back to you.
Thanks and get back to you soon,
Best
Iolanda
Hi Steve (@Vernalis),
sorry I completely forgot to write you back. In the end I managed to solve. So the problem was due to a single structure which in the initial milion subset was written in the wrong way:
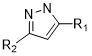
The nitrogen of the pyrazole ring was missing its hydrogen and thus 3D coordinates were not generated for one molecule and this was responsible for the error in the PMI node.
So all good and the workflow works just fine.
Thanks for your support
Best regards
Iolanda
Hi,
Thanks for getting back to me. Could I ask you, what was in the input cell for this molecule? Was it a missing/empty cell or something else - it would be good to know to see what broke it and see if we can make it more resistant!
Steve
Hi Steve,
Yes I was actually getting back to you on this. The cell on the 3D coordinates was empty. I just made a trial for you to show you what is visible:
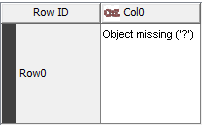
You can also try to reproduce yourself, try for example C1(C2=CC=CC=C2)=N[N]C(C3CCC3)=C1
The thing is that the 3D coordinate will proceed as normal, so it will simply leave the empty cell for those molecules which do not work, but the node itself will make its work. It is the PMI that will fail. I think a way to go around it is to add an option in the PMI node to “ignore empty cells” or something like that. I tried to put a node in between the 3D coordinates node and the PMI code to split the table and remove the rows with empty cell from the 3D coordinates but somehow it didn’t work… no sure if I was doing something wrong though.
Hope it helps, let me know if you need any other info,
Best
Iolanda
Ah, OK - thank you. That’s definitely a bug in the node. Hopefully not too difficult to fix and I can get it out in our next update.
Steve
Sounds good, thanks for your responsiveness all the way through  .
.
Best
Iolanda
Hi @Iolanda,
Thanks for this test molecule:
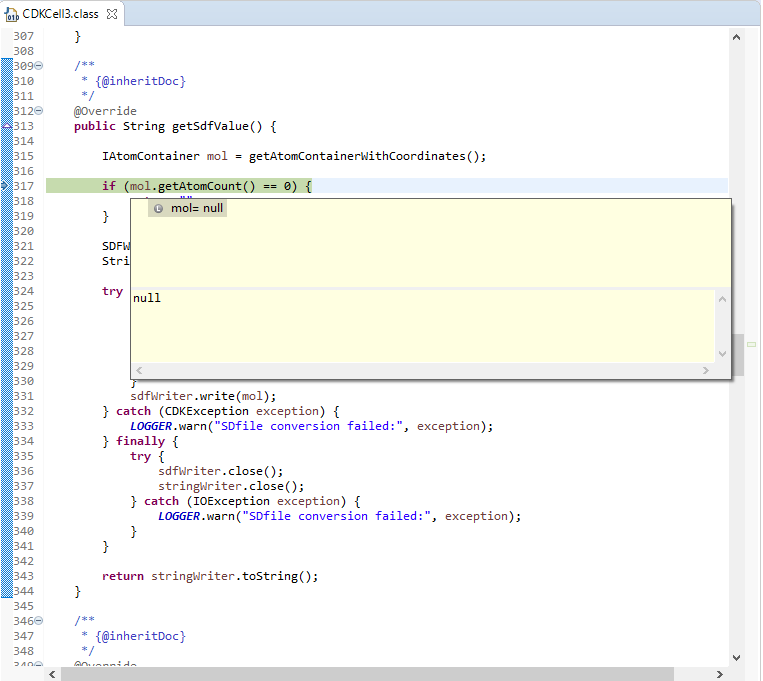
I’ve managed to track down the problem which is actually in the CDK plugin:
It seems that 3D Coordinates node does not output a proper ‘Missing Cell’, but instead a CDKCell with null molecule, which then breaks when you attempt to get the SDF block from it. I’m not sure who the maintainers of the CDK plugin are, so I’m going to tag @thor and @gab1one here to see if they can put us in touch with them.
We’ve not come across this behaviour having not tried to use the CDK nodes previously. Incidentally, the Molecule to CDK node thinks it can parse that molecule OK, but leaves out any input rows with missing values, which was also confusing behaviour.
EDIT: Some further digging reveals that the problem is the CDK toolkit believes it can process the above input, but it’s deserialization step fails to retrieve it. In particular the SmilesParser call in line 125 here throws a SmilesParseException:
Steve
To the best of my knowledge, the forum user @egonw is the contact for the CDK nodes.
btw you can take a look at the cdk nodes sources here: https://github.com/cdk/nodes4knime
@Iolanda - if you want to make it work with CDK, then if you use a CDK to Molecule node before the PMI node then that will replace the molecule it cannot read with a missing cell and all is ‘well’ (well, nearly!)
Steve
Hi @Vernalis Steve, happy to hear that the whole story was useful to identify that issue!
I got the impression that the problem was on the way how the cell was “labeled” as also the filtering of missing values didn’t work. Ok all good! And thank you for tip on putting a “molecule node” in between!
Best
Iolanda
Yes, basically in this case, when the CDK Renderer tries to render the cell contents it can’t read the molecule back in from the store and displays a ‘?’, which happens to look very like a normal Missing Value!
I think it is probably actually a problem deep within the CDK toolkit rather than KNIME itself. The KNIME CDK Cell stores a molecule by writing the SMILES String representation followed by various add-ons (co-ordinates, atom types, bond colouring etc). During coordinate generation, explicit H’s are added (but not to the c1nncc1 ring, as that could be a radical as implied!), and once they are present the written SMILES then is rejected.
Steve
Thanks Steve (@Vernalis) ! Yes all clear now! I managed to do all calculations on my dataset, however now I threw the ChEMBL database in the workflow, filtered 100K structures and did all the rest. The 3D coordinates works but again it has found some structures that it is not able to convert into a 3D space. So I have tried to introduce the CDK to molecule node but it doesn’t seem to solve the problem.
I understand that it is all a matters of the CDK nodes so I shouldn’t probably continue to bother you Maybe @egonw can you look at that?
The alternative is to use one of the RDKit-based conformation nodes:
or from the Vernalis nodes:
(But yes, it would be really nice if the CDK nodes worked too!)
Steve
Perfect Steve, I will give them a look!
Thank you!!
This topic was automatically closed 90 days after the last reply. New replies are no longer allowed.