I’m trying the align-it pharmacophore generator node of the 3d-e-chem silico-it package. In principle this node can be really useful for the cheminformatics community and represent a great addition to KNIME but I encountered some oddity that would need to be clarified.
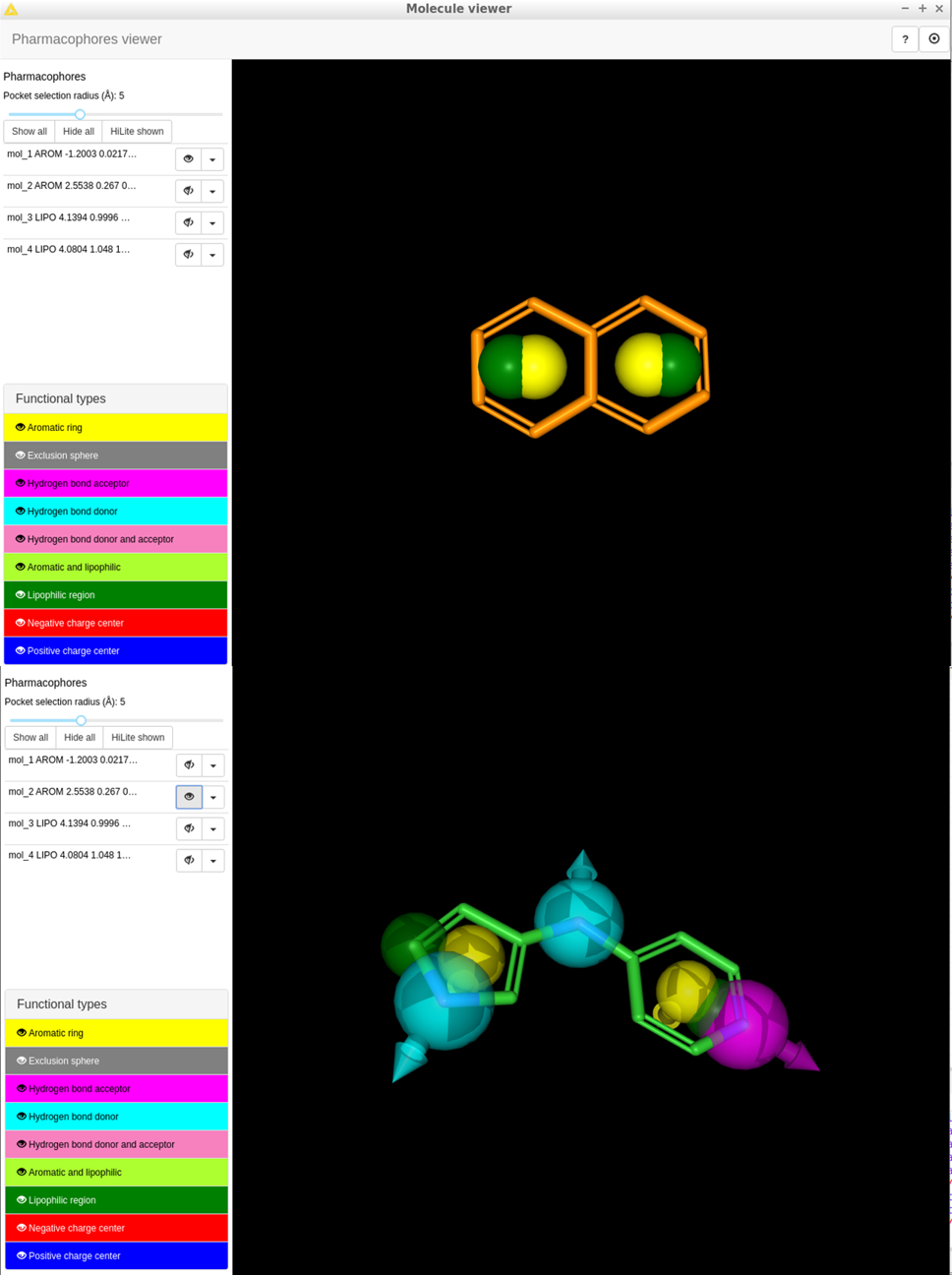
In the attached workflow (91.1 KB) I sketched 4 simple molecules, 3 of them used in the official align-it documentation to explain the pharmacophore generation process. You can see that the generated pharamacophores are different from what expected (see figures).
In general the following problem appears:
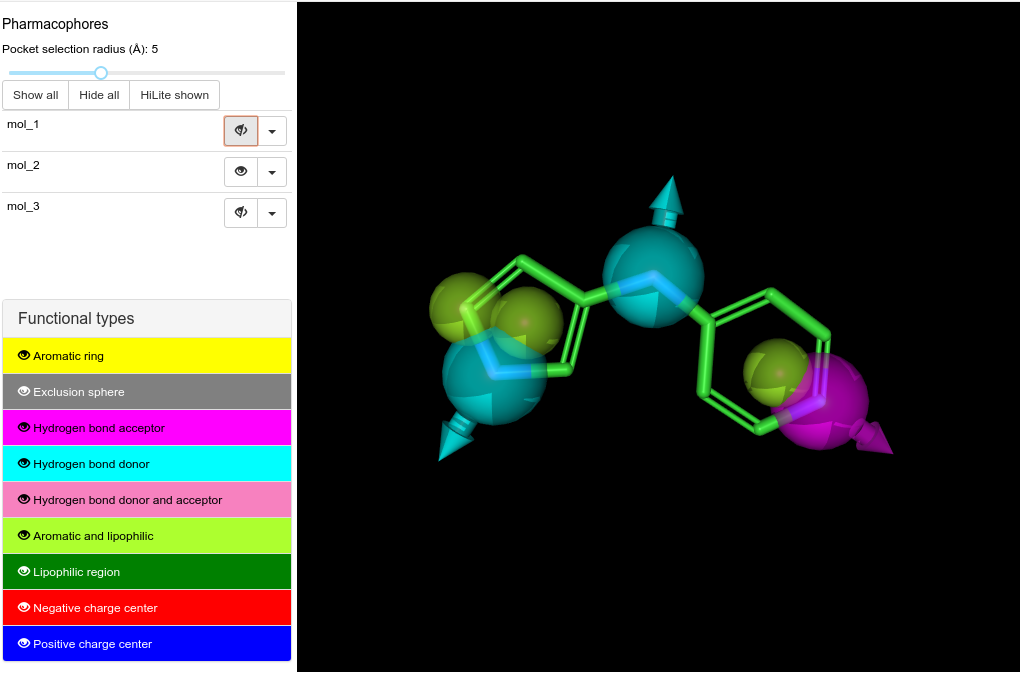
No pure aromatic nor lipophilic features are created. Instead are generated always mixed aromatic/lipophilic features also in non-aromatic moyeties (see mol_3 figure).
Mixed aromatic/lipophilic features center seem to not overlap with aromatic/aliphatic ring centers. May this be due to the pharmacophore points fusing process?
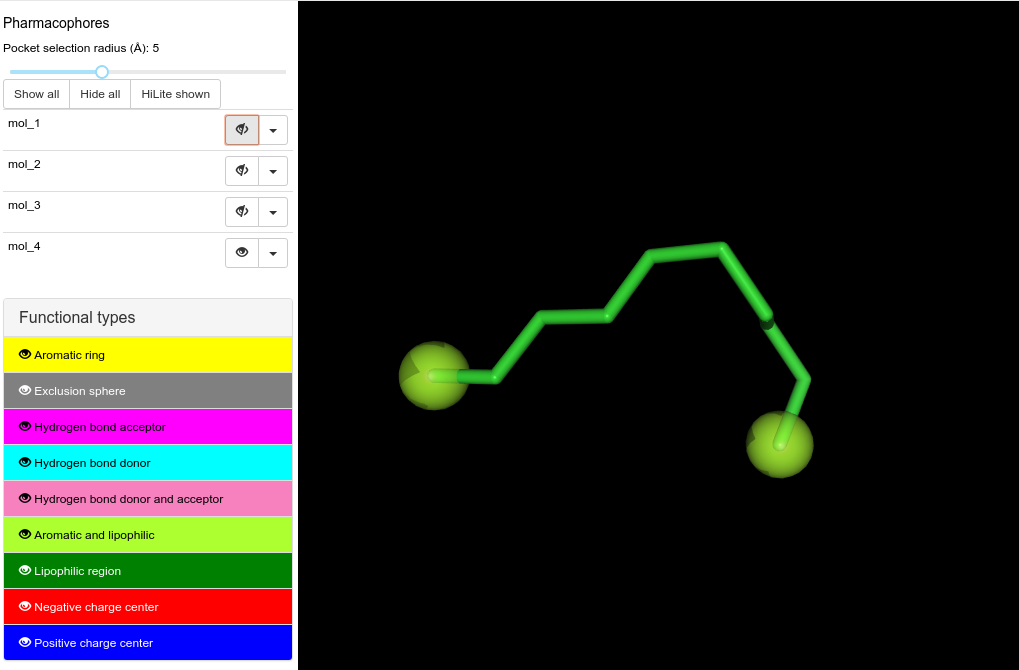
No lipophilic features are created on aliphatic chains (see mol_3 and mol_4)
A mixed aromatic/lipophilic feature appeared centered on a ring atom but not fused with the aromatic/lipophilic feature of the ring (see mol_2)
I think this is a question of default align-it behaviour, which seems to be to provide hybrid pharmacophore points. The java snippet node in the align-it Pharmacophore Generator meta-node provides all the arguments to passed to align-it, so this can be edited to get the behaviour I think you’re after. A simple modification under //Enter your code here: would be
Thank you very much for your help. I appreciated it.
So, from this results it seems that the hybrid functionality is created with an “aromatic OR lipophilic” feature instead that an “aromatic AND lipophilic” one, as appear in the viewer. From that I misunderstood the concept.
It remains to be understood why the last molecule (i.e. mol_4, a lipophilic chain) gives 2 lipophilic features at both ends of the molecules but not along the chain, while mol_2 has one lipophilic one on an aromatic atom.
Hmm. I see the lipophilic points for Mol 3 are not as they should be according to (schematic) figure 4b in the documentation too. The two lipohilic pharmacophore points in the propyl group are not centred on the terminal atoms, while the two lipophilic points on the long alkyl chain (mol 4) are centred exactly on the terminal atoms. Looks like the two isopropyl group lipophiles could have been affected by a merge, but the alkyl chain lipophiles remain puzzling. The middle ethyl chain in Mol 3 should also form a lipophilic group, but does not.
The textual description of the rules for joining the lipohilic centres is perhaps unclear. Other molecules drawn with substitutions seem to give more plausible results, but any(?) number of linear acyclic CH2 groups get ignored/merged to nothing unless attached to a CHn where n≠2. Not sure what the expected behaviour should be, but the documentation says there is a threshold for contribution to a lippphilic group of 9.87, equivalent to half the contribution of an exposed methyl carbon terminating a chain. Seems likely to exclude the CH2 groups, but I’m not rash enough to call it a bug/feature without more thought… Other pharmacophores not in the original WF were checked e.g. for +ve centres are picked up OK.